Water Structures and Packing Efficiency in Methylene Blue Cyanometallate Salts




1
EMAT, Department of Physics, University of Antwerp, Groenenborgerlaan 171, 2020 Antwerp, Belgium
2
Dipartimento di Scienze Chimiche, della Vita e della Sostenibilità Ambientale, Università degli Studi di Parma, Via Parco Area delle Scienze 17/A, 43124 Parma, Italy
*
Authors to whom correspondence should be addressed.
Crystals 2020, 10(7), 558; https://doi.org/10.3390/cryst10070558
Submission received: 26 May 2020
/
Revised: 4 June 2020
/
Accepted: 5 June 2020
/
Published: 1 July 2020
(This article belongs to the Special Issue A 10 Years Journey: Chemical, Physical, and Biological Properties and Applications of Crystals)
Abstract
:Crystal structure prediction is the holy grail of crystal engineering and is key to its ambition of driving the formation of solids based on the selection of their molecular constituents. However, this noble quest is hampered by the limited predictability of the incorporation of solvent molecules, first and foremost the ubiquitous water. In this context, we herein report the structure of four methylene blue cyanometallate phases, where anions with various shapes and charges influence the packing motif and lead to the formation of differently hydrated structures. Importantly, water molecules are observed to play various roles as isolated fillings, dimers, or an infinite network with up to 13 water molecules per repeating unit. Each crystal structure has been determined by single-crystal X-ray diffraction and evaluated with the aid of Hirshfeld surface analysis, focussing on the role of water molecules and the hierarchy of different classes of interactions in the overall supramolecular landscape of the crystals. Finally, the collected pieces of evidence are matched together to highlight the leading role of MB stacking and to derive an explanation for the observed hydration diversity based on the structural role of water molecules in the crystal architecture.
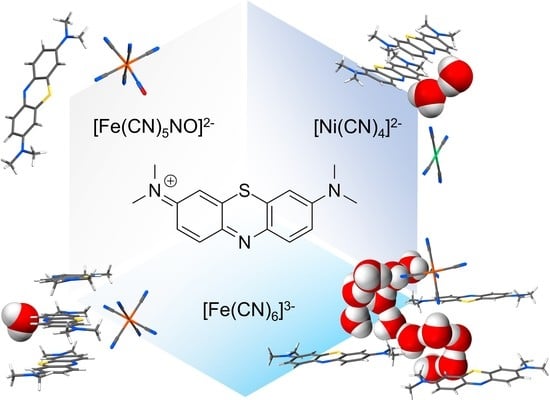
1. Introduction
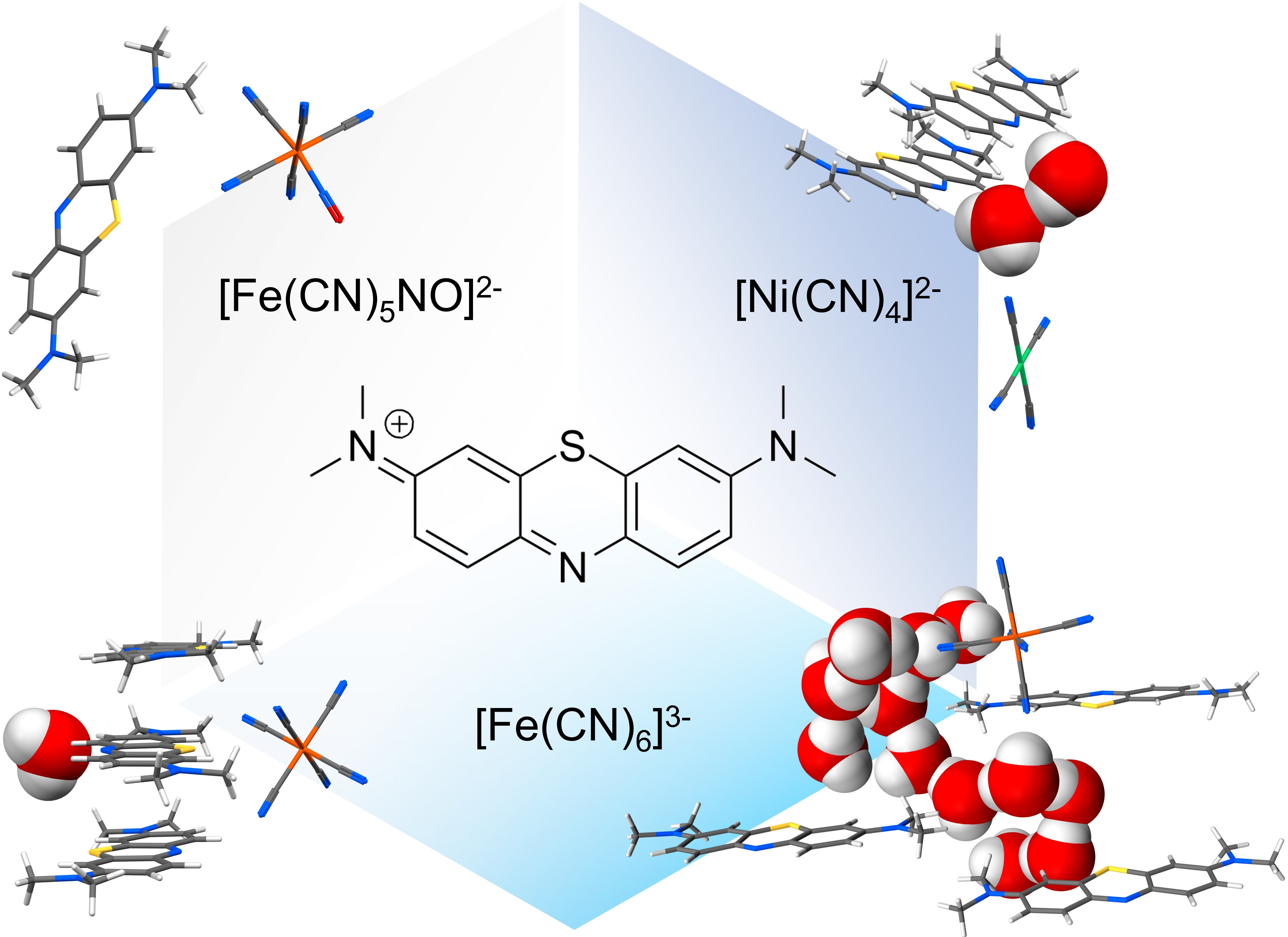
Methylene blue (MB) is a widely known compound whose redox and optical properties have been studied and employed for decades in chemistry [1,2], medicine [3,4,5,6], and biology [7,8]. Commonly commercialised as a chloride salt, MB owes its most relevant properties to the cationic heteroaromatic compound 3,7-bis(dimethylamino)-phenothiazin-5-ium (MB+; chemical formula C16H18SN3+ as shown in Figure 1a), which is responsible for the intense blue colour of its aqueous solutions. As far as its solid-state behaviour is concerned, two interactions play a dominant role: the aromatic π-π stacking between MB+ and its same neighbours, and the electrostatic and hydrogen bond interactions involving the lone pair of its aromatic nitrogen. This last propensity is determined by the peculiar molecular electronic structure of MB+ and particularly by the presence of an exposed σ lone pair on this nitrogen, as discussed in our previous study [9]. Both these interaction tendencies are recognizable in the interaction probability map of the isolated cation, shown in Figure 1b [10]. While on the one hand the importance of aromatic stacking in the crystal packing of MB+ is supported by its occurrence in all the structures reported up until now [11,12], on the other hand various stacking modes have been reported depending on the anionic counterpart and the presence of other species, especially solvents, participating in the crystal architecture [13]. In this regard, methylene blue salts have shown a strong tendency to incorporate water molecules in their periodic structures, not rarely with various possible stoichiometries for a single type of counter-anion.
Perhaps the most representative example is the pseudo-polymorphism of methylene blue hydrates, of which up to five distinct forms with different amounts of structural water molecules have been reported [14]. In particular, the occurrence of such hydration polymorphism is known to complicate crystal structure prediction and synthesis of molecular solids to the point that water has been regarded to as the “nemesis of crystal engineering” [15]. This makes systematic studies on the role of water in the crystal packing of molecular materials particularly valuable and functional to the definition of strategies to identify the main aspects ruling the formation of water-based pseudo-polymorphism. Aiming to conduct such investigation on the crystallisation processes of methylene blue salts, we synthesized a series of four methylene blue cyanometallate phases using anions with different charges and geometries, and in the presence of water traces in the crystallisation media (non-anhydrous organic solvents). The choice of this class of metal complexes allowed us to selectively vary the geometric and charge features of the anionic moieties, while maintaining the type of chemical groups that they contribute with to the overall supramolecular network. For each phase, the overall packing features are scrutinised to understand how water molecules fit into the interactions landscape, and finally identify the primary reasons for their presence and solid-state interactions.
2. Materials and Methods
2.1. General Remarks
All solid and liquid reagents have been purchased by commercial sources and used without further purification. Crystallisations by slow solvent evaporation were conducted at room temperature and pressure, and in the presence of air and atmospheric moisture.
2.2. Synthetic Procedures
2.2.1. Synthesis of MB2[Ni(CN)4]·2H2O
Fifty-one milligrams of methylene blue pentahydrate (0.159 mmol) and 19 mg of K2Ni(CN)4 (0.079 mmol) were transferred into a 25 mL glass vial. Twenty millilitres of N,N-dimethylformamide was added to the powder mixture and the vial was closed with a screw cap and transferred into an ultrasound bath to be sonicated for 5 min. The resulting clear blue solution was left to evaporate in an open 50 mL crystallisation dish. After 7 days of slow solvent evaporation, dark metallic-green needles were found in solution, and were subsequently isolated for single-crystal X-ray analysis.
2.2.2. Synthesis of MB2[Fe(CN)5NO]
Ten milligrams of methylene blue pentahydrate (0.03 mmol) and 5 mg of Na2[Fe(CN)5NO] (0.015 mmol) were transferred into a 25 mL glass vial, and 20 mL of a H2O-acetone mixture 1:1 was added to the powder mixture and the vial was closed with a screw cap and transferred into an ultrasound bath to be sonicated for 5 min. The thus obtained clear blue solution was left to evaporate in a 50 mL crystallisation dish. After 20 days of slow evaporation, golden needle-shaped crystals were found and subsequently collected for single-crystal X-ray analysis.
2.2.3. Synthesis of MB3[Fe(CN)6]·H2O and MB3[Fe(CN)6]·12.36H2O
Twenty milligrams of methylene blue pentahydrate (0.06 mmol) and 7 mg of K3Fe(CN)6 (0.02 mmol) were transferred into a 25 mL glass vial. Then, 20 mL of N,N-dimethylformamide was added to the powder mixture and the vial was closed with a screw cap and transferred into an ultrasound bath to be sonicated for 5 min. The resulting clear blue solution was left to evaporate in an open 50 mL crystallisation dish. After 6 days of slow solvent evaporation, two crystalline phases were observed present in the mother liquor: golden needles and metallic dark-green prisms. Both crystal phases were isolated for single-crystal X-ray analysis.
2.3. Single-Crystal X-ray Diffraction Analysis
Crystallographic analyses on single crystal specimens of MB2[Fe(CN)5NO], MB3[Fe(CN)6]·H2O and MB3[Fe(CN)6]·12.36H2O were carried out at the XRD1 beamline at the Elettra synchrotron (CNR Trieste) [16]. Diffraction data were collected at 100K using a monochromatic 0.7 Å wavelength (17.712 keV), using a cold nitrogen stream produced with an Oxford Cryostream 700 (Oxford Cryosystems Ltd., Oxford, UK). Diffraction images were recorded on a Pilatus 2M detector. Diffraction data on MB2[Ni(CN)4]·2H2O were collected on a SMART APEX2 diffractometer using graphite monochromated Mo Kα radiation (λ = 0.71073 Å) and an APEX II CCD detector.
Synchrotron datasets were processed using the software package Rigaku CrysAlisPro ver. 1.171.40.67a [17]. Empirical absorption correction was performed by the SCALE3 ABSPACK program ver. 1.0.11 included in the CrysAlisPro software suite. Diffraction data of MB2[Ni(CN)4]·2H2O were processed using the software Bruker Apex3 [18], performing an empirical “multi-scan” absorption correction. All the structure determinations were conducted using Olex2 software ver. 1.3.0 [19], using the programs ShelXT and ShelXL for structure solution and least square refinement of the atomic coordinates, respectively. All non-hydrogen atoms were refined anisotropically. Suitable restraints assisted the anisotropic refinement of atoms belonging to disordered molecules. All hydrogen atoms were located upon examination of residual electron densities in the difference Fourier maps and therefore placed and fixed in idealized positions. Further details of data reduction, crystallographic refinements for each sample and reciprocal space sections of the disordered phases MB2[Fe(CN)5NO] and MB3[Fe(CN)6]·H2O are provided in the supplementary materials.
The structures reported in this work can be accessed free of charge from the Cambridge Structural Database website (https://www.ccdc.cam.ac.uk/structures/); deposition numbers 2004494–2004497.
2.4. Computational Details
Hirshfeld surface calculation functional to the creation of Hirshfeld plots used in the discussion was conducted by using the software Crystal Explorer version 17.05 [20]. Each species was set to singlet state, and molecular charges were adjusted prior to the calculation. This was performed using the default settings (surface: “Hirshfeld”, property: “none”) and “very high” resolution settings.
Atomic packing factors (APFs) were calculated as APF = 1 − Vvoid/Vcell. The Vvoid parameters were derived by using the “Display Void” function of the software “Mercury” of the Cambridge Structural Database System [11]. For this calculation, the “contact surface” method was used with a probe radius of 0.2 Å and a grid spacing of 0.1 Å. Disordered structures have been considered in only one of their equivalent configurations. Their crystallographic symmetry was reduced to the pure translational symmetry (P1 settings) by using the software Vesta [21] and the excess atoms were removed by the model, resulting in a chemically sound structure, which was used for the APF calculations.
3. Results and Discussion
3.1. Systematic Variation of Cyanometallate Ions
The effect of both charge and geometry of the anions were probed by employing three different species: the square planar tetracyanonickelate(II) [Ni(CN)4]2−, the octahedral nitroprusside [Fe(CN)5(NO)]2− and the octahedral ferricyanide [Fe(CN)6]3−. Given the isoelectronic character of cyanide and nitrosyl groups and therefore of the two iron anions, their use combined with the divalent nickel species allows us to test the effects of different charge ([Fe(CN)5(NO)]2− vs. [Fe(CN)6]3−), different geometry ([Ni(CN)4]2− vs. [Fe(CN)5(NO)]2−) or both combined ([Ni(CN)4]2− vs. [Fe(CN)6]3−). By using these anions, four products were obtained as the use of ferricyanide resulted in two distinct phases with significantly different water content. Interestingly, all four products show a unique crystal packing situation not only concerning methylene blue cations’ stacking and overall degree of solvation, but also with respect to structural disorder. These aspects are described below for each product. A detailed description of the crystallographic modelling can be found in the supplementary materials.
3.2. Crystal Structure Analysis
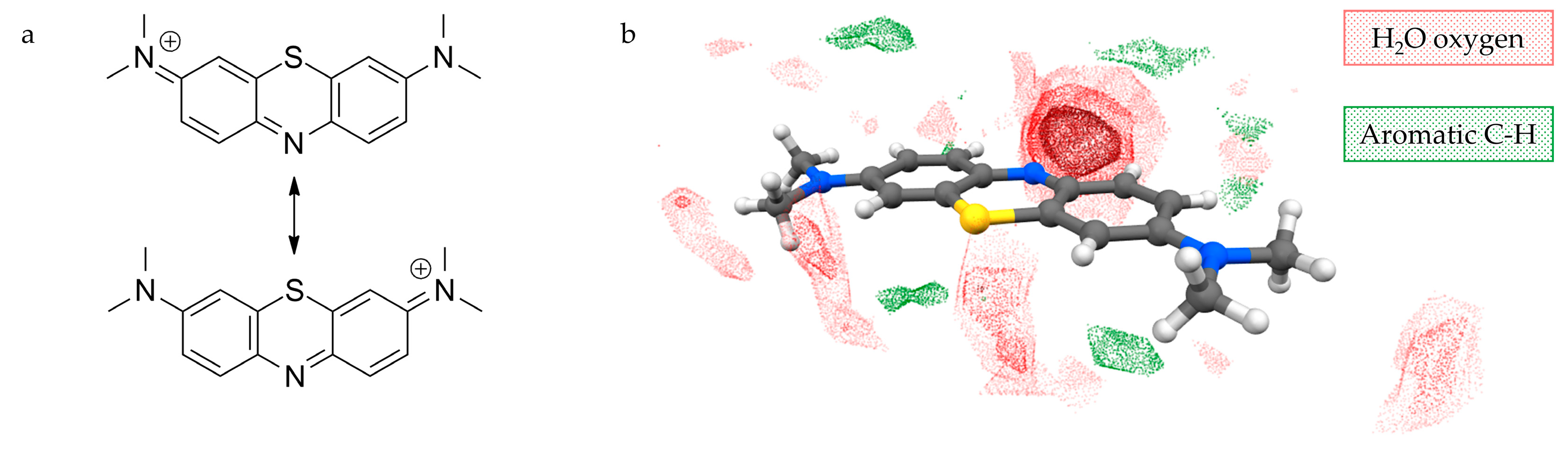
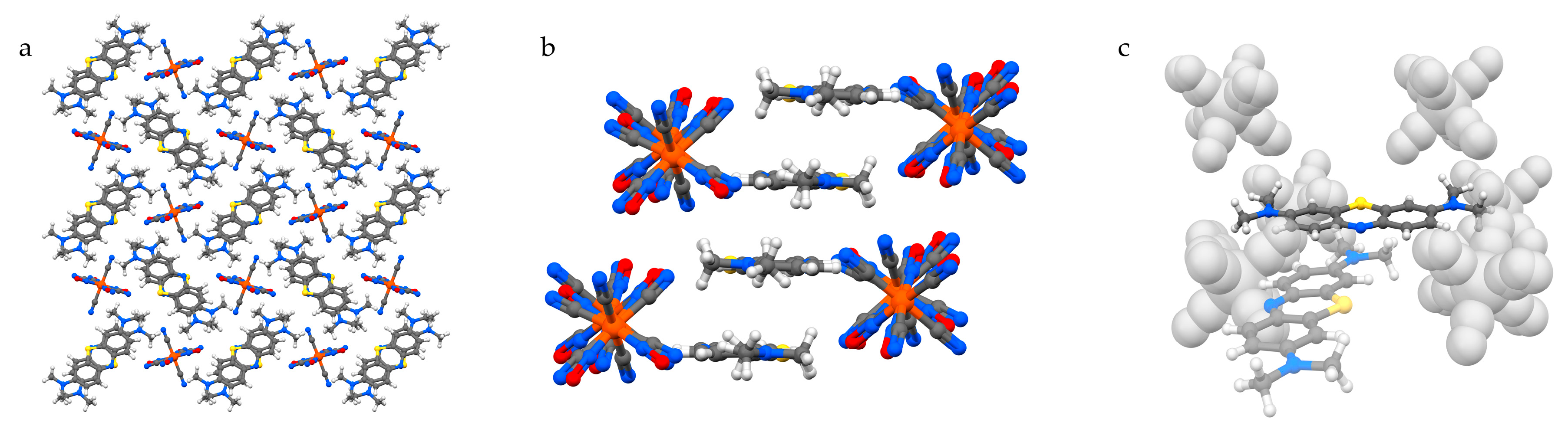
The structure of MB2[Ni(CN)4]·2H2O (1) features MB cations performing infinite zig-zag stacking columns separated by vertically aligned anions, and connected by water-based hydrogen bonds (Figure 2). The mutual orientation of the MB cations in the stacking columns consists of two types, which we define as opposite (O) or same (S), and alternates forming dimers of same-orientation MB+ (Figure 2a,b). The resulting sequence along the stacking direction is “…O–S–O–S…”, as can be observed in Figure 2b. The presence of one water molecule for each MB ensures the stabilisation of the aromatic nitrogen lone pair via hydrogen bonding (Figure 2c; distances: 2.197(16) Å for H···N1 and 2.224(15) Å for H···N2). Additional hydrogen bonds with the methyl groups from MB belonging to different columns and with cyanonickelate ions allow every pair of water molecules to connect three distinct MB+ columns and one anion. It is worth highlighting that the stacking columns share the same alignment of MB molecules, which lie with their longer dimension along the [1 0 0] direction. The flat cyanonickelate ions also show this orientation. Although the role of the solvent in stabilising the observed configuration should not be underestimated, the described packing preference might be strongly promoted by the presence of flat anions, which can act as spacers between MB+ columns without hindering their formation.
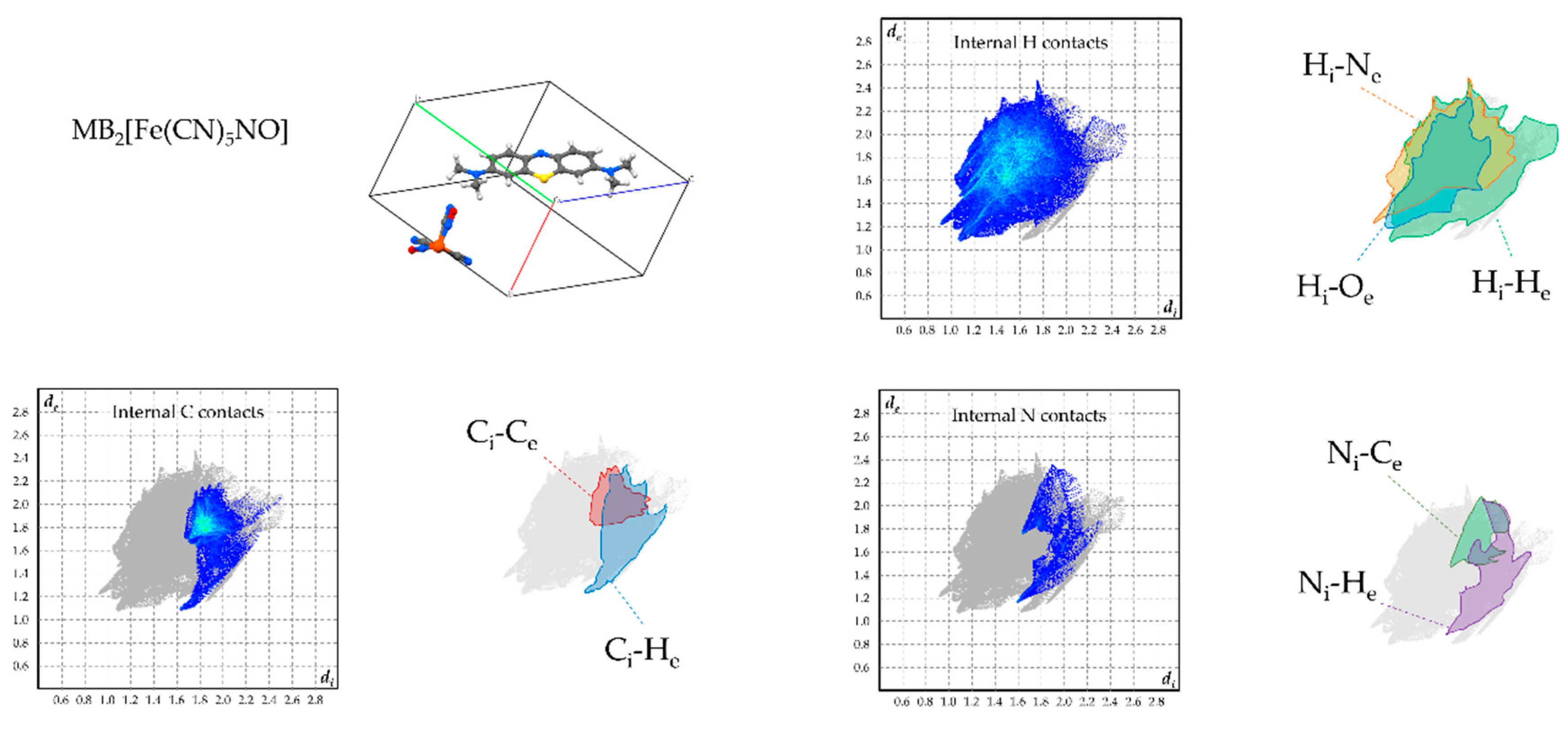
In MB2[Fe(CN)5NO] (2), the system copes with a substantially different dimensionality of the anion. As a result, MB+ columns are not aligned in a common direction, but lie orthogonal to each other, creating square channels bordered with methyl groups. Here, the anions are piled up and accept hydrogen bonds from the methyl groups onto every cyanide/nitrosyl ligand (Figure 3a). As every MB+ contributes to four different “anionic” channels and along the stacking direction the length covered by one anion corresponds to that of a MB+ dimer, the cations adopt an “opposite” stacking type to yield a more favourable hydrogen-bonding coverage for the anions (Figure 3b). These latter anions lie in sites having Ci symmetry and show positional disorder where the nitrosyl ligand is alternative to the cyanide groups in four coplanar positions. Importantly, no water molecules participate in the crystal, and the closest hydrogen bond donor for the MB aromatic nitrogen is a methyl group belonging to an adjacent column, although the CH···N distance of 2.871(2) Å highlights its rather weak character (Figure 3c).
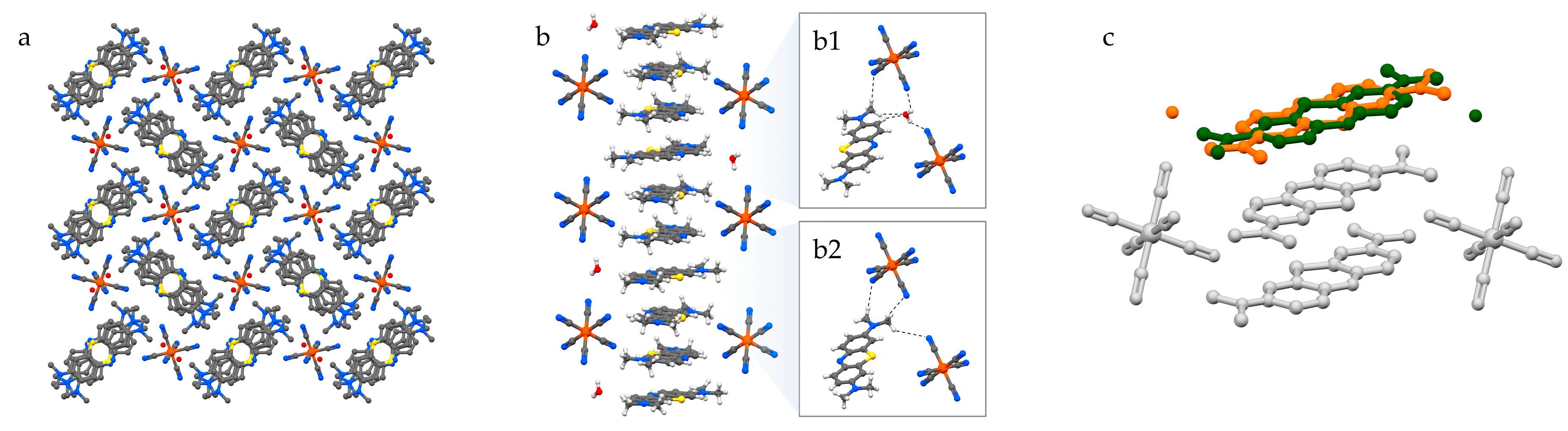
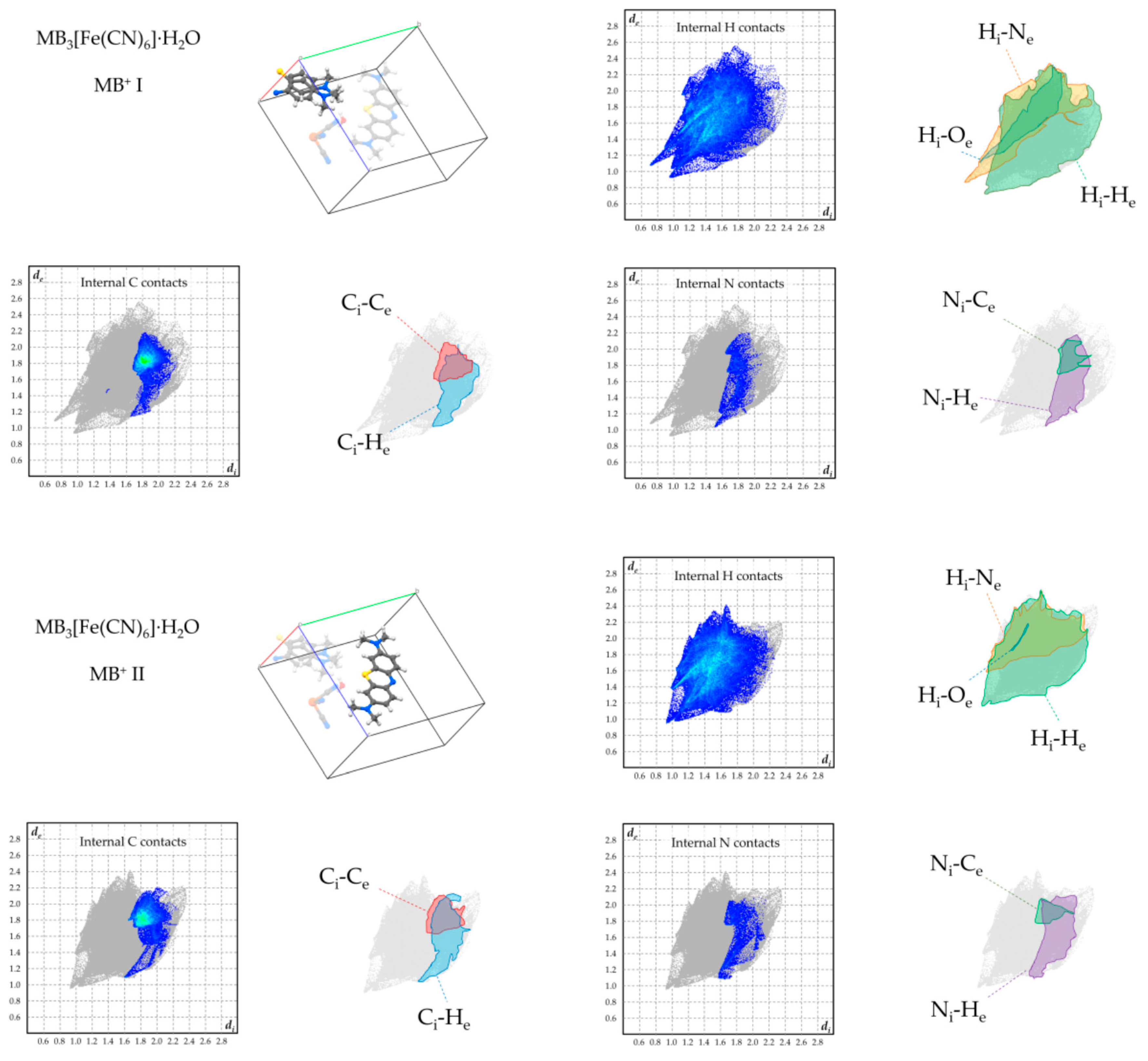
A similar arrangement is found in MB3[Fe(CN)6]·H2O (3), where likewise stacked MB columns create square channels to allocate ferricyanide anions (Figure 4a). However, in this case, the trivalent anion is distributed differently along the channel due to the charge requirements. In fact, while in 2 each MB pair is surrounded by four anions and each anion is embraced by four MB pairs—hence the charge balance—in the case of 3, the distribution of the anion along the channels is such that every anion in embraced by four MB+ stacked trimers. Since, as discussed above, the thickness of the anion matches that of a MB+ dimer, residual spaces remain in the anion-filled channels. Half of these gaps are filled by having one water molecule accepting a double hydrogen bond by the aromatic and aliphatic CH groups of MB+ and donating hydrogen bonds to anions above and below (Figure 4(b1)). The remaining gaps are occupied by a MB+ methyl group located on the opposite side with respect to the water molecule, and donating hydrogen bonds to the surrounding anions in a similar manner as water molecules (Figure 4(b2)). Since the gaps filled by water and those filled by the methyl group are symmetry-equivalent, the [MB+·H2O] units are found to be disordered in the solid phase. Consequently, in the MB+ stacking column, every ordered pair of molecules is preceded and followed by a disordered [MB+·H2O] unit (Figure 4c).
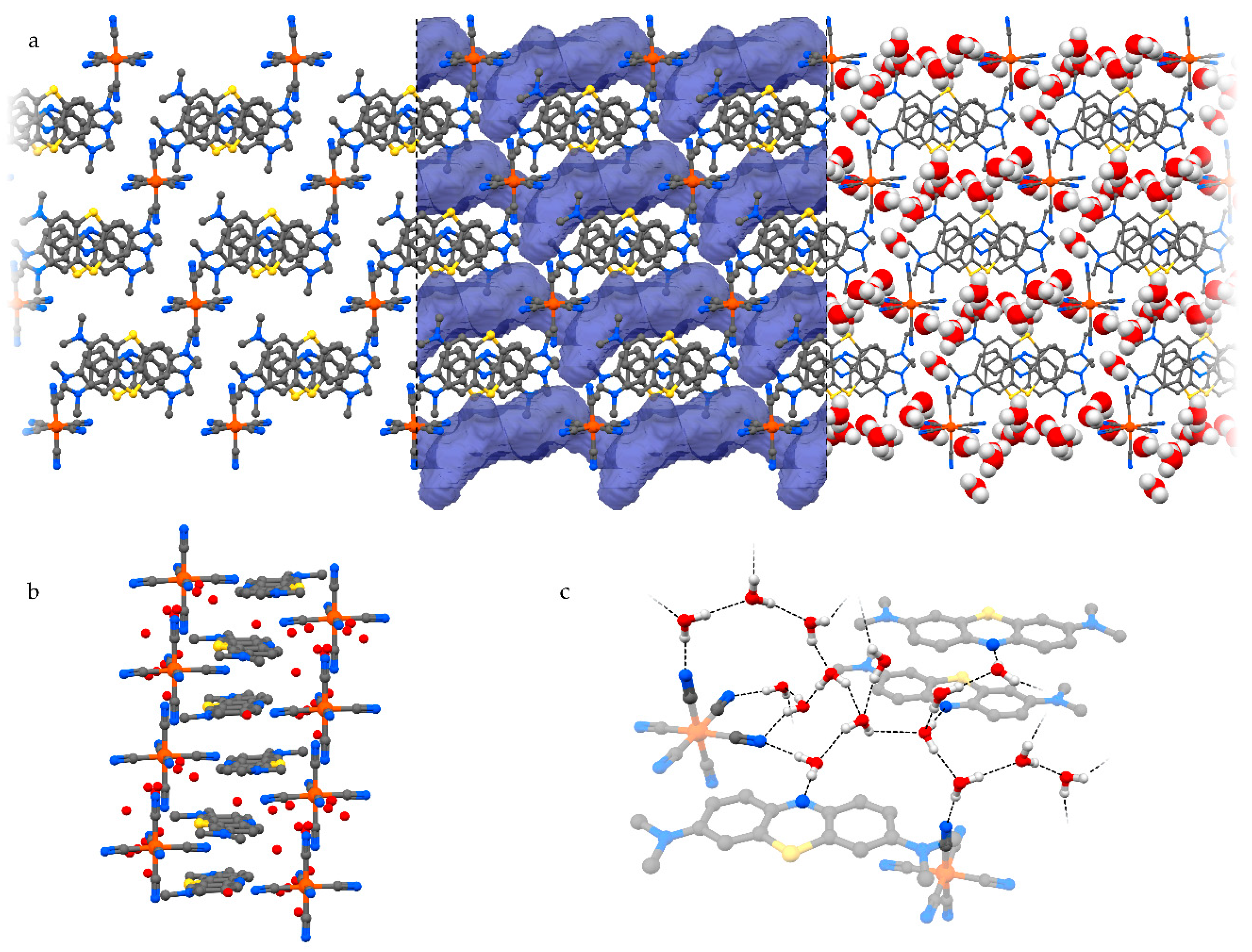
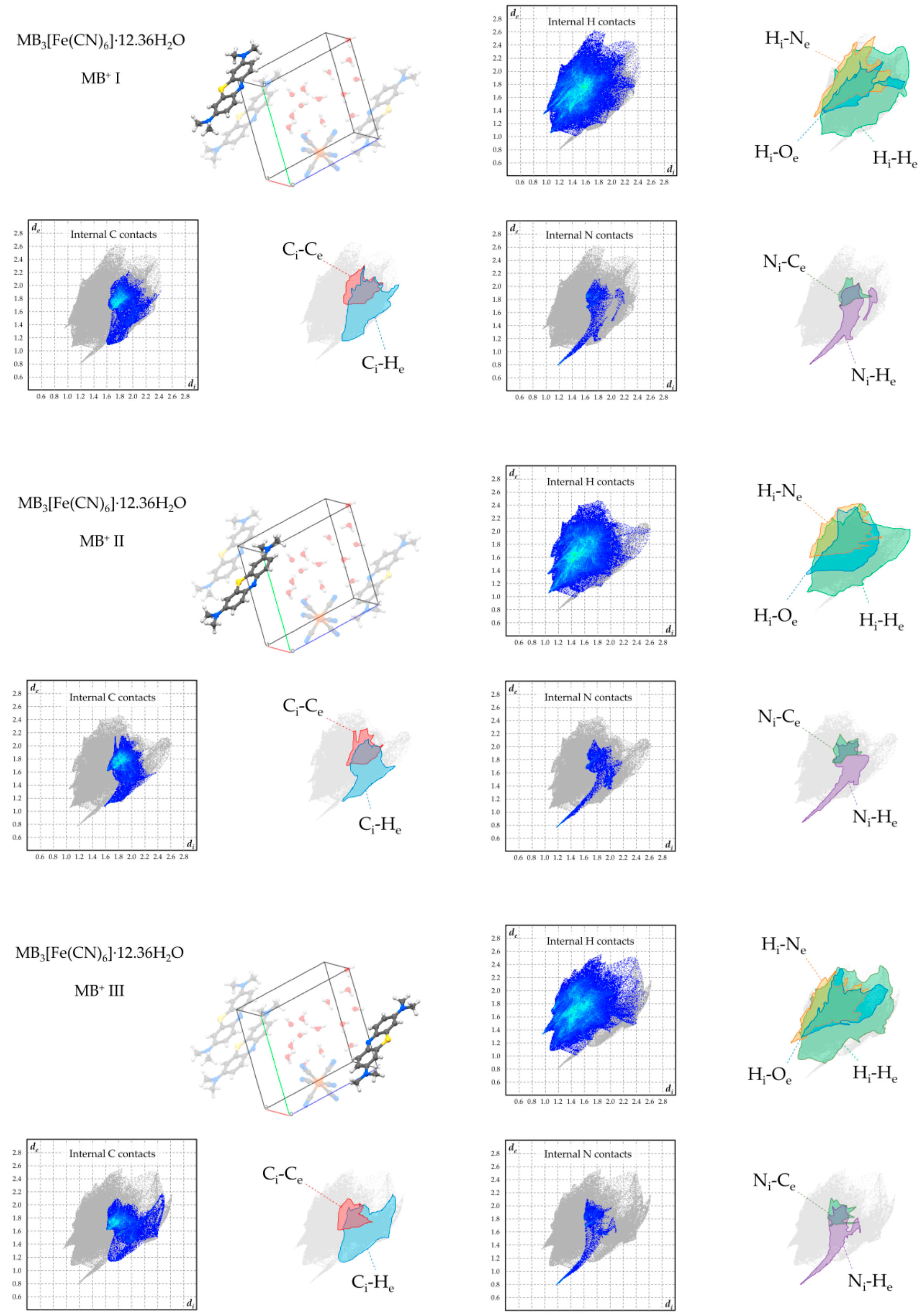
The presence of orientationally disordered [MB+·H2O] units is not the only observed solution for an efficient crystal packing of MB+ with ferricyanide ions. Indeed, the same synthesis that yielded crystals of 3 also produced a second phase, where for every anion there are up to thirteen independent water molecules (Figure 5). These perform a complex network of hydrogen bonds along the [0 0 1] direction, which connects all the ions in the solid phase and saturates the aromatic nitrogen lone pair of every MB+ (Figure 5c). Two of these water molecules are partially substituted with a single one in an intermediate position, resulting in the unit cell formula MB3[Fe(CN)6]·12.36H2O (4). The isolation of the anionic species by such a large amount of solvent molecules and the flexibility of the latter in adapting the network of hydrogen bonding to the packing needs of the remaining species allow MB+ to perform perfectly ordered stacked columns (Figure 5b).
3.3. Packing Considerations
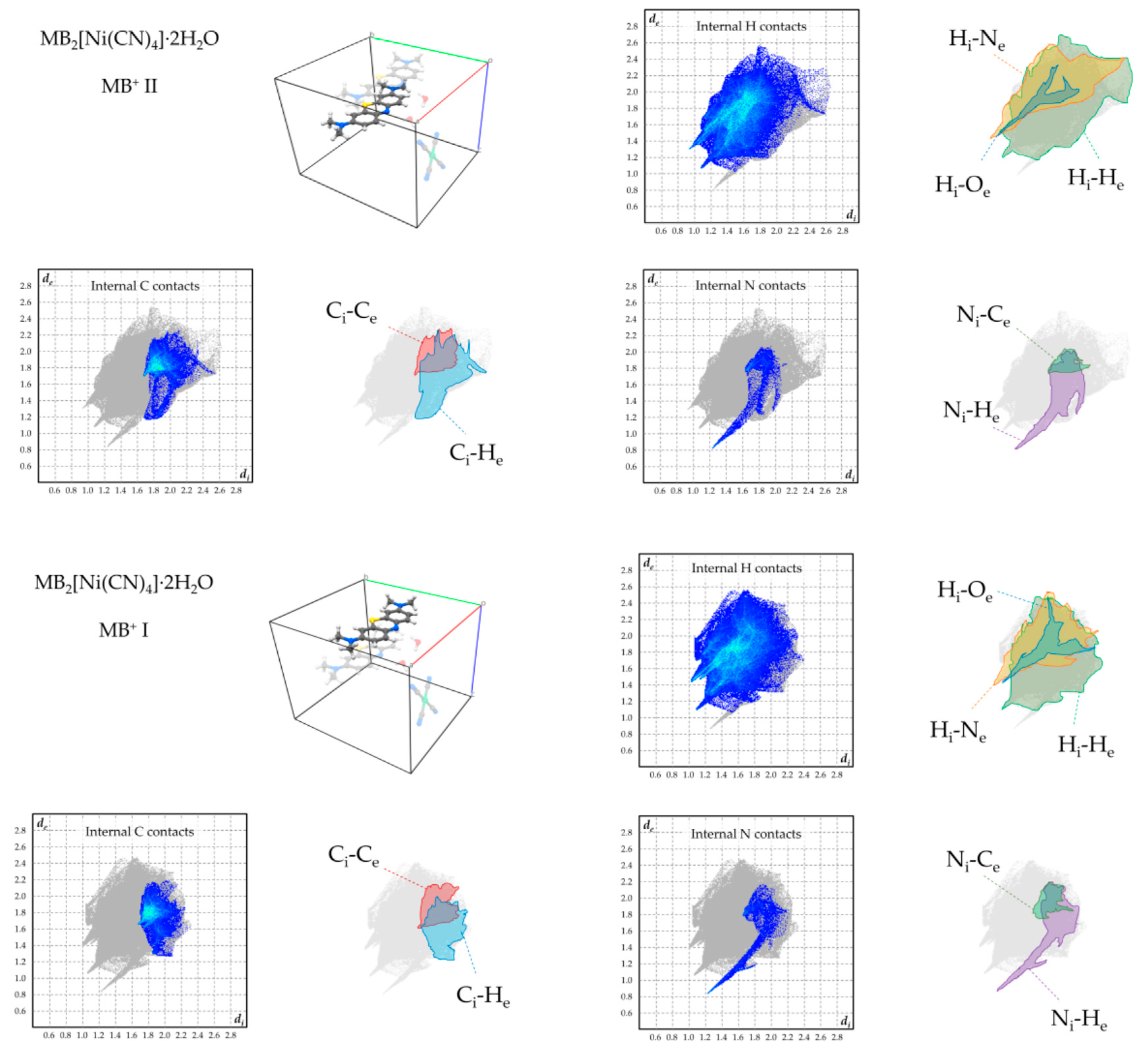
To overview the supramolecular-interaction environment of MB+ in the obtained phases, Hirshfeld fingerprint plots were computed for each symmetry-independent cation, focussing on the contacts involving its hydrogen, carbon, and nitrogen atoms (Figure 6, Figure 7, Figure 8 and Figure 9). [22,23]. In all products, the strong tendency of MB+ to form stacked columns is further evidenced by the presence, in the Hirshfeld plot, of characteristic C–C contact isles centred at approximately de = di = 1.8Å (highlighted in red in the figures). The shape of this region is nearly symmetric with respect to the de = di axis, hence spotlighting an overall homogeneity of the stacking geometry and distance involving distinct MB molecules. In particular, a perfectly symmetric C–C contact distribution is present in the case of 2, which is an obvious consequence of having only one MB per asymmetric unit and therefore only a single MB-MB stacking geometry is possible. As far as finer details on this matter are concerned, every phase shows a singular combination of O or S type of MB stacking. Precisely, the stacking sequence repeating units for every phase are [–O–S–] for 1, [–O–] for 2, [–O–S/O–] for 3 and [–O–S–O–] for 4. Accordingly, the least symmetric Ci–Ce contact distributions between MB+ species are found for phases 1 and 4, which have periodic sequences of both S and O types, while the MB orientational disorder present in 3 averages out the differences between these stacking types, resulting in a much less evident Ci–Ce distributions asymmetricity. As clearly displayed in the Hirshfeld plots—especially those relative to H and N contacts—the supramolecular interaction landscape of MB is different and strongly asymmetric in each phase due to the various molecules involved. One very distinguishable trait, which is only exhibited by compounds 1 and 4, is the presence of hydrogen bonds with the aromatic nitrogen of MB+. This feature is evidenced by the sharp spike in the lower half of the “Internal N contacts” plot in Figure 5 and Figure 8. In these two phases, every MB+ accepts such interactions from water molecules, while in phases 2 and 3, the absence or relatively scarce amount of crystallisation solvent leaves the lone pair of the heteroatom without significant interactions.
A tentative explanation for the formation of such structures even when water molecules are present in the crystallisation environment could be hypothesized upon considering the atomic packing factors (APFs) of phases 1–4 (Table 1). APFs are generally defined as the ratio between the volume occupied by all atoms in the unit cell and the unit cell volume. These values are strongly influenced by directional interactions such as hydrogen bonds, which prioritise the energy stabilisation they provide over the overall packing efficiency, as in the case of the renowned porous structure of hexagonal ice [24]. In this regard, it is widely acknowledged in the crystal engineering community that a better crystal packing is not always synonymous with a higher occurrence likelihood when predicting the structure of polymorphic phases [25]. In our cases, the lowest APF is found for 1 (0.731), whereas 2 and 3 have remarkably better packing efficiency (APF 0.752 and 0.765, respectively). On the one hand, this difference also originates, to a large extent, from the packing motif due to the anion geometry. On the other hand, the comparison between 3 and 4 (APF value: 0.754) highlights uniquely the effects of a different phase hydration. This is observed to increase the void fraction of the phase by ~ 1.1% when water molecules participate in the packing and saturate with hydrogen bonds every lone pair present on the central nitrogen atoms of MB.
In summary, our results suggest that, when water molecules are present in the crystallisation medium, their participation in the crystal lattice of MB metallate salts can be driven by the need for stabilising the aromatic nitrogen lone pair. However, this need has a lower priority than optimising the infinite stacking of the cation, and when the latter requirement matches with a high packing coefficient, as in case of 2 and 3, the stabilisation of the aromatic nitrogen can be sacrificed. Compound 3 constitutes a special instance in this regard, as the non-optimal packing required a water molecule to fill the small gaps, without any possibility of hydrogen bonding with the MB+ aromatic nitrogen. The paramount priority of MB stacking among supramolecular interactions is further evidenced by considering two structures reported earlier by our group: MB chloride dihydrate and MB bisulfate [13]. These two cases exhibit the same packing motif, space group and analogous unit cell parameters as, respectively, compound 1 and 2. In the case of MB bisulfate, hydrogen-bonded dimers of the anions with an overall charge of −2 occupy the same channels where in compound 2 the nitroprusside anions are located. This suggests that the shape, volume, and charge of the anion—or, in the cited case, of its dimer—generally play a more relevant role than its internal chemical structure. This aspect ceases to be relevant in the cases of chloride and tetracyanonickelate ions, which have totally different charge, shape, and volume, and yet lead to the formation of two iso-packed phases, therefore leaving MB+ infinite stacking columns as the main leading packing prerequisite.
In conclusion, our study highlights how the challenge of predicting the formation of hydrate phases can be addressed by identifying and ranking the supramolecular interactions needs of the involved species. Its validity should not be limited to molecular salts but may also be considered for other non-ionic products of crystal engineering such as co-crystals and supramolecular assemblies in general. In fact, taking into consideration both packing efficacy and the requirements of specific types of interactions of the species involved, combinatorial studies of the type we have shown can spotlight hierarchies in the energy landscape of crystal phases, which can assist in the prediction of the presence and role of water molecules in the overall supramolecular architecture.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4352/10/7/558/s1, additional details on the single-crystal XRD analysis of the described compounds can be found in the supplementary materials, available free of charge on the publisher’s website.
Author Contributions
All authors contributed significantly to this work. Further details are reported as follows. Synthesis: S.C., D.C.; crystallographic analysis: S.C., C.G.; supervision: G.P., C.G., manuscript writing and graphics: S.C. All authors have read and agreed to the published version of the manuscript.
Acknowledgments
The Elettra Synchrotron (CNR Trieste) is gratefully acknowledged for the beamtime allocated at the beamline XRD1 (proposal nr 20175216). S.C. acknowledges the Research Foundation Flanders (FWO) for supporting his research (grant nr. 12ZV120N).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kalaitzakis, D.; Kouridaki, A.; Noutsias, D.; Montagnon, T.; Vassilikogiannakis, G. Methylene Blue as a Photosensitizer and Redox Agent: Synthesis of 5-Hydroxy-1 H -pyrrol-2(5 H )-ones from Furans. Angew. Chem. Int. Ed. 2015, 54, 6283–6287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Begum, R.; Najeeb, J.; Sattar, A.; Naseem, K.; Irfan, A.; Al-Sehemi, A.G.; Farooqi, Z.H. Chemical reduction of methylene blue in the presence of nanocatalysts: A critical review. Rev. Chem. Eng. 2019. [Google Scholar] [CrossRef]
- Schirmer, R.H.; Adler, H.; Pickhardt, M.; Mandelkow, E. “Lest we forget you - methylene blue…”. Neurobiol. Aging 2011, 32, 2325.e7–2325.e16. [Google Scholar] [CrossRef] [PubMed]
- Ginimuge, P.R.; Jyothi, S.D. Methylene blue: Revisited. J. Anaesthesiol. Clin. Pharmacol. 2010, 26, 517–520. [Google Scholar] [PubMed]
- Lo, J.C.Y.; Darracq, M.A.; Clark, R.F. A review of methylene blue treatment for cardiovascular collapse. Can. J. Emergency Med. Canadian 2014, 46, 670–679. [Google Scholar] [CrossRef]
- Oz, M.; Lorke, D.E.; Hasan, M.; Petroianu, G.A. Cellular and molecular actions of Methylene Blue in the nervous system. Med. Res. Rev. 2011, 31, 93–117. [Google Scholar] [CrossRef] [Green Version]
- Gutter, B.; Speck, W.T.; Rosenkranz, H.S. A study of the photoinduced mutagenicity of methylene blue. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 1977, 44, 177–181. [Google Scholar] [CrossRef]
- Li, R.; Chen, J.; Cesario, T.C.; Wang, X.; Yuan, J.S.; Rentzepis, P.M. Synergistic reaction of silver nitrate, silver nanoparticles, and methylene blue against bacteria. Proc. Natl. Acad. Sci. USA 2016, 113, 13612–13617. [Google Scholar] [CrossRef] [Green Version]
- Canossa, S.; Bacchi, A.; Graiff, C.; Pelagatti, P.; Predieri, G.; Ienco, A.; Manca, G.; Mealli, C. Hierarchy of Supramolecular Arrangements and Building Blocks: Inverted Paradigm of Crystal Engineering in the Unprecedented Metal Coordination of Methylene Blue. Inorg. Chem. Front. 2017, 56, 3512–3516. [Google Scholar] [CrossRef]
- Wood, P.A.; Olsson, T.S.G.; Cole, J.C.; Cottrell, S.J.; Feeder, N.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Evaluation of molecular crystal structures using Full Interaction Maps. Cryst. Eng. Comm. 2013, 15, 65–72. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge structural database. Acta. Crystallogr. B. Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Cambridge Structural Database System, Version 5.41; Cambridge Crystallographic Data Centre: Cambridge, UK, 2020.
- Canossa, S.; Predieri, G.; Graiff, C. Hydrogen bonds and π-π Interactions in two new crystalline phases of methylene blue. Acta Crystallogr. Sect. E Crystallogr. Commun. 2018, 74, 587–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rager, T.; Geoffroy, A.; Hilfiker, R.; Storey, J.M.D. The crystalline state of methylene blue: A zoo of hydrates. Phys. Chem. Chem. Phys. 2012, 14, 8074–8082. [Google Scholar] [CrossRef] [PubMed]
- Clarke, H.D.; Arora, K.K.; Bass, H.; Kavuru, P.; Ong, T.T.; Pujari, T.; Wojtas, L.; Zaworotko, M.J. Structure-stability relationships in cocrystal hydrates: Does the promiscuity of water make crystalline hydrates the nemesis of crystal engineering? Cryst. Growth Des. 2010, 10, 2152–2167. [Google Scholar] [CrossRef]
- Lausi, A.; Polentarutti, M.; Onesti, S.; Plaisier, J.R.; Busetto, E.; Bais, G.; Barba, L.; Cassetta, A.; Campi, G.; Lamba, D.; et al. Status of the crystallography beamlines at Elettra. Eur. Phys. J. Plus 2015, 130, 1–8. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction. CrysAlisPro Software System, Version 1.171.40.67a; Rigaku Oxford Diffraction: Oxford, UK, 2017.
- Bruker AXS. APEX3 Software, Version 2016.1-0; Bruker AXS: Madison, WI, USA, 2016.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Wolff, S.K.; Grimwood, D.J.; McKinnon, J.J.; Turner, M.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer, Version 17.05; University of Western Australia: Perth, Australia, 2012.
- Momma, K.; Izumi, F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld surface analysis. Cryst. Eng. Comm. 2009, 11, 19–32. [Google Scholar] [CrossRef]
- McKinnon, J.J.; Spackman, M.A.; Mitchell, A.S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr. Sect. B Struct. Sci 2004, 60, 627–668. [Google Scholar] [CrossRef]
- Durão, J.; Gales, L. Permeation of Light Gases through Hexagonal Ice. Materials 2012, 5, 1593–1601. [Google Scholar] [CrossRef]
- Corpinot, M.K.; Bučar, D.K. A Practical Guide to the Design of Molecular Crystals. Cryst. Growth Des. 2019, 19, 1426–1453. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Resonant structures of MB+ (a) and its interaction probability map (b) calculated by using the “Full Interaction Map” tool of the software Mercury [10]. As in all the other figures, the light elements colour code is: C = grey; O = red; S = yellow; N = blue; H = white.
Figure 1.
Resonant structures of MB+ (a) and its interaction probability map (b) calculated by using the “Full Interaction Map” tool of the software Mercury [10]. As in all the other figures, the light elements colour code is: C = grey; O = red; S = yellow; N = blue; H = white.

Figure 2.
Crystal packing of 1 viewed along the [0 0 1] and [1 0 0] crystal directions (part “a” and “b” respectively). The hydrogen bonding network allowed by the water molecules is shown in part “c”.
Figure 2.
Crystal packing of 1 viewed along the [0 0 1] and [1 0 0] crystal directions (part “a” and “b” respectively). The hydrogen bonding network allowed by the water molecules is shown in part “c”.

Figure 3.
Crystal packing of 2 viewed along the [1 0 0] and [−1 1 1] crystal directions (part “a” and “b” respectively). In “c”, the chemical environment of MB+ in the asymmetric unit is displayed without showing the MB+ stacked below and above and by showing the anion in white for the sake of clarity.
Figure 3.
Crystal packing of 2 viewed along the [1 0 0] and [−1 1 1] crystal directions (part “a” and “b” respectively). In “c”, the chemical environment of MB+ in the asymmetric unit is displayed without showing the MB+ stacked below and above and by showing the anion in white for the sake of clarity.

Figure 4.
Crystal packing of 3 viewed along the [1 0 0] and [0 1 1] crystal directions (part “a” and “b” respectively). In “c”, the two parts of the disordered [MB+·H2O] unit are highlighted in green and orange, while the ordered species are displayed in white.
Figure 4.
Crystal packing of 3 viewed along the [1 0 0] and [0 1 1] crystal directions (part “a” and “b” respectively). In “c”, the two parts of the disordered [MB+·H2O] unit are highlighted in green and orange, while the ordered species are displayed in white.

Figure 5.
(a) Crystal packing of 4 viewed along the [0 0 1] direction and displayed, sequentially, without water molecules, with the voids they occupy, and in their presence. (b) Stacking of MB+, where the hydrogen atoms are not displayed for the sake of clarity. (c) Unit cell content highlighting the water molecule network, focusing on the dodeca-hydrate type of unit cell. The first three molecules from the left and the last three on the right are related by translational symmetry.
Figure 5.
(a) Crystal packing of 4 viewed along the [0 0 1] direction and displayed, sequentially, without water molecules, with the voids they occupy, and in their presence. (b) Stacking of MB+, where the hydrogen atoms are not displayed for the sake of clarity. (c) Unit cell content highlighting the water molecule network, focusing on the dodeca-hydrate type of unit cell. The first three molecules from the left and the last three on the right are related by translational symmetry.

Figure 6.
Hirshfeld plots of relevant supramolecular contacts of MB+ in 1. The subscripts “i” and “e” means “internal” or “external”, respectively, with respect to the considered molecule.
Figure 6.
Hirshfeld plots of relevant supramolecular contacts of MB+ in 1. The subscripts “i” and “e” means “internal” or “external”, respectively, with respect to the considered molecule.

Figure 7.
Hirshfeld plots of relevant supramolecular contacts of MB+ in 2.

Figure 8.
Hirshfeld plots of relevant supramolecular contacts of MB+ in 3.

Figure 9.
Hirshfeld plots of relevant supramolecular contacts of MB+ in 4.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Void volume, percentage and atomic packing factors (APFs) of the obtained phases.
| Phase | Void Volume (Å3) | Void Fraction (%) | Atomic Packing Factor |
|---|---|---|---|
| MB2[Ni(CN)4]·2H2O | 980.74 | 26.9 | 0.731 |
| MB2[Fe(CN)5NO] | 445.26 | 24.8 | 0.752 |
| MB3[Fe(CN)6]·H2O | 585.17 | 23.5 | 0.765 |
| MB3[Fe(CN)6]·12.36H2O | 378.07 | 24.6 | 0.754 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Canossa, S.; Graiff, C.; Crocco, D.; Predieri, G. Water Structures and Packing Efficiency in Methylene Blue Cyanometallate Salts. Crystals 2020, 10, 558. https://doi.org/10.3390/cryst10070558
AMA Style
Canossa S, Graiff C, Crocco D, Predieri G. Water Structures and Packing Efficiency in Methylene Blue Cyanometallate Salts. Crystals. 2020; 10(7):558. https://doi.org/10.3390/cryst10070558
Chicago/Turabian StyleCanossa, Stefano, Claudia Graiff, Domenico Crocco, and Giovanni Predieri. 2020. "Water Structures and Packing Efficiency in Methylene Blue Cyanometallate Salts" Crystals 10, no. 7: 558. https://doi.org/10.3390/cryst10070558
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.