PdI2 as a Simple and Efficient Catalyst for the Hydroamination of Arylacetylenes with Anilines

 ,
,  ,
, 
Abstract
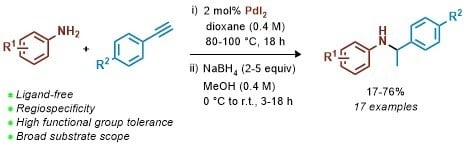
:
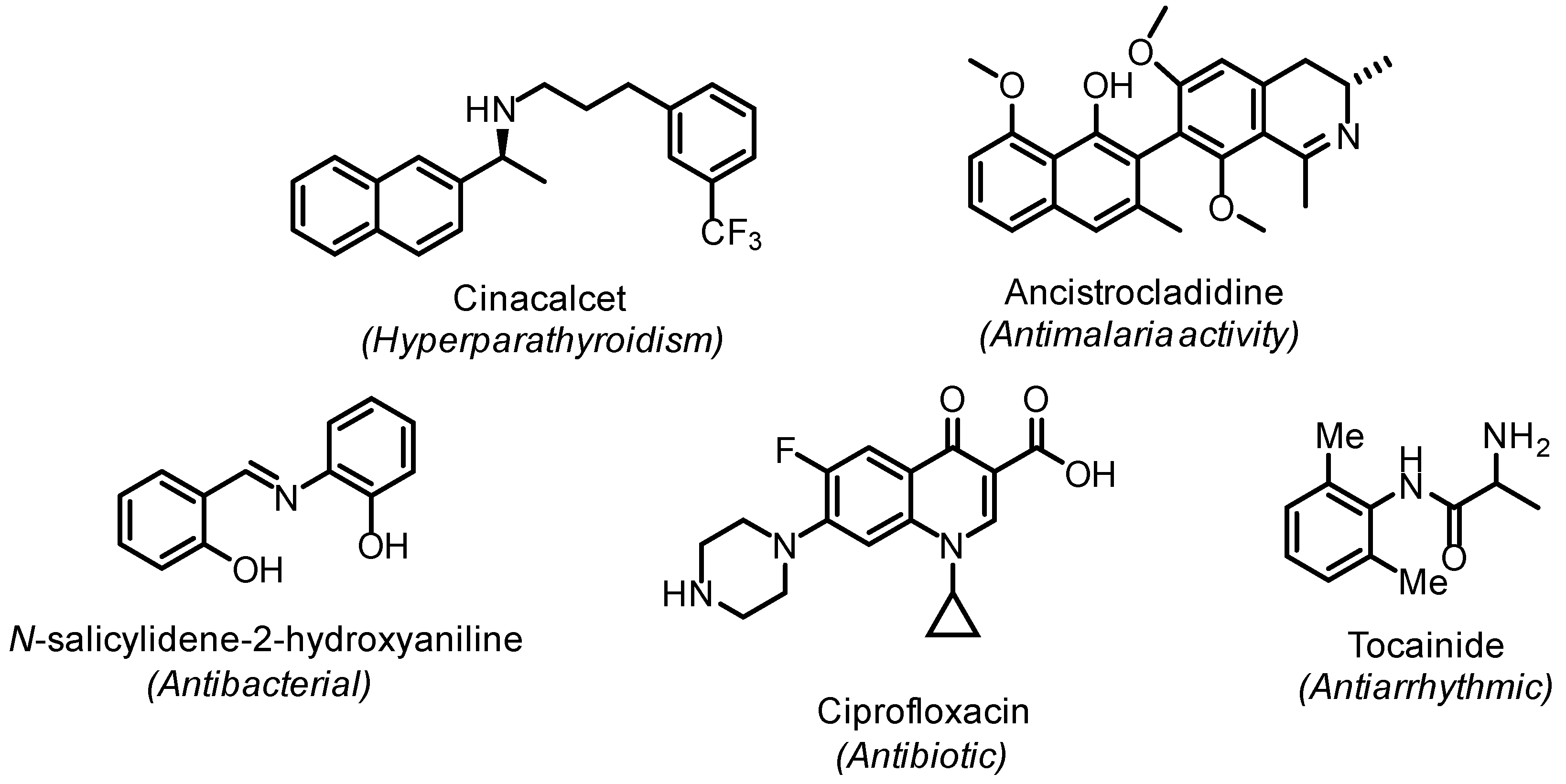
1. Introduction
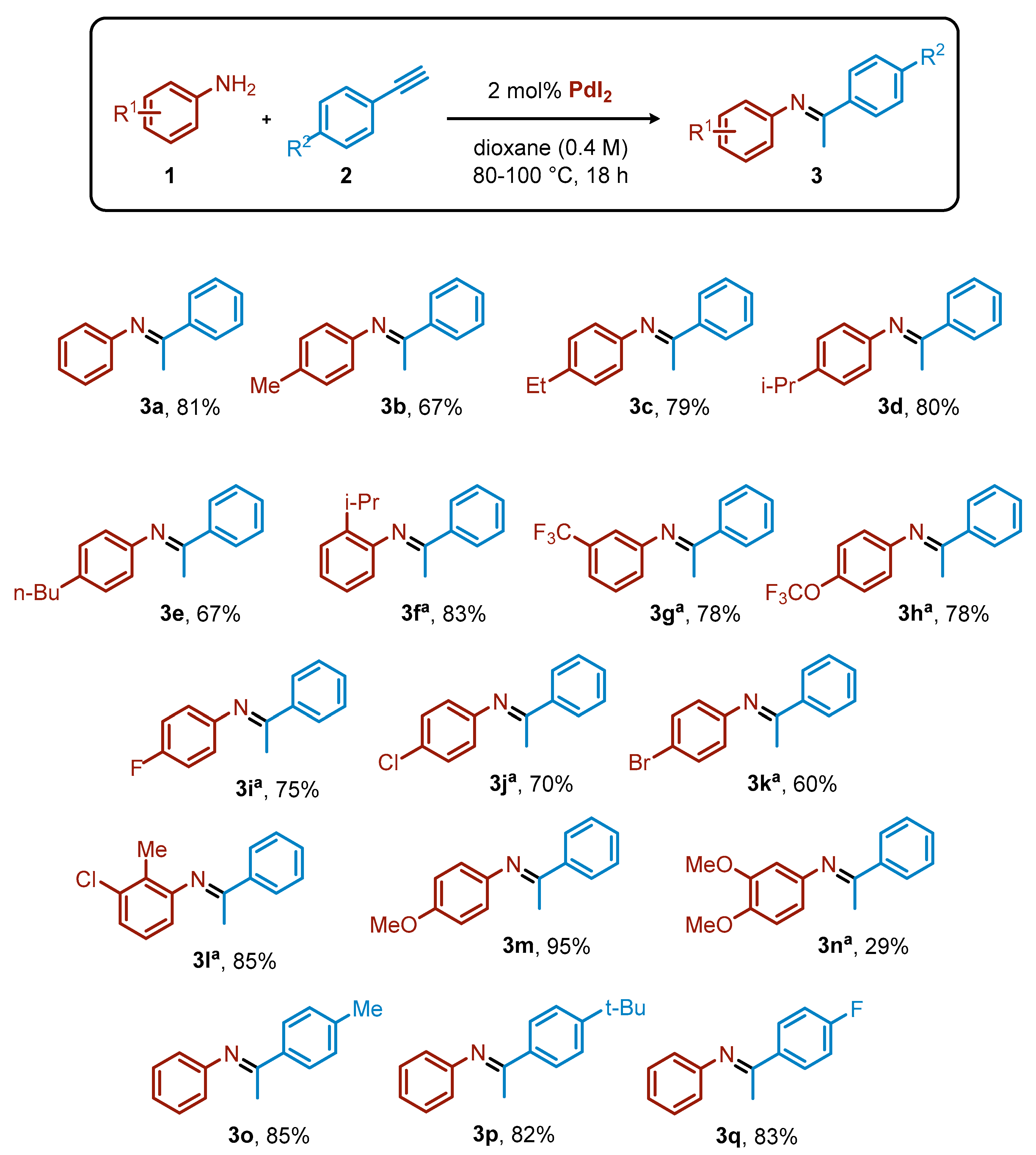
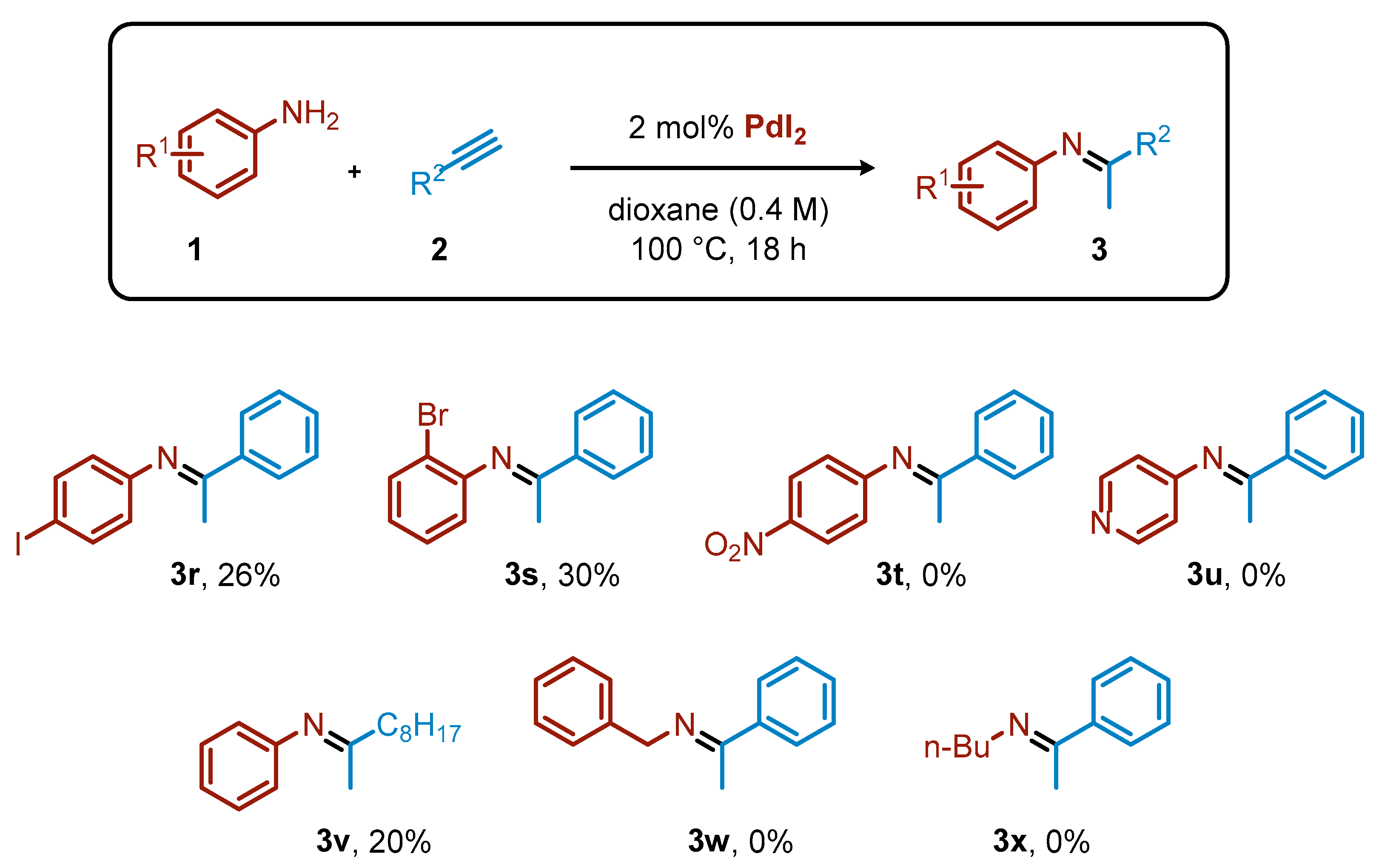
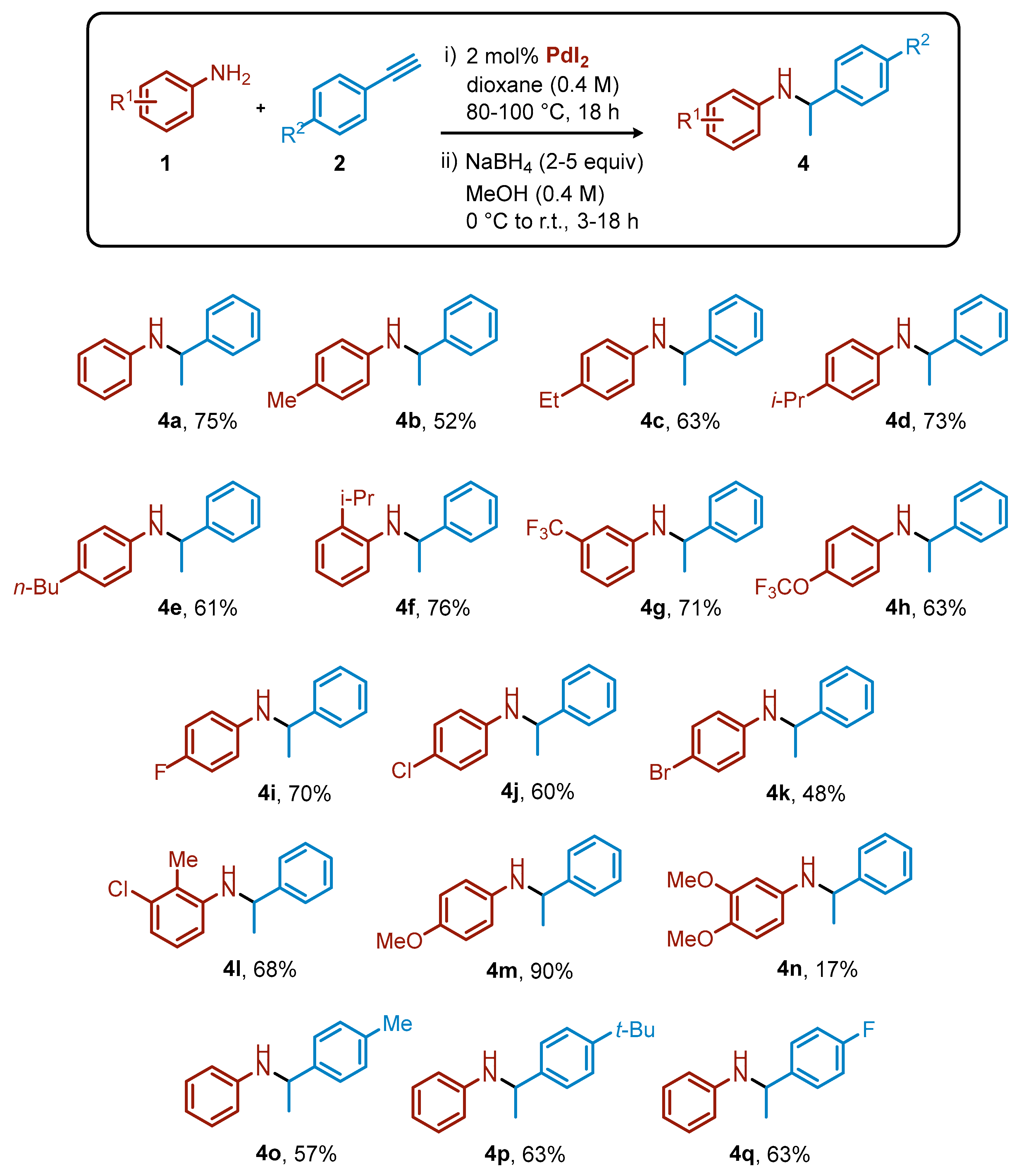
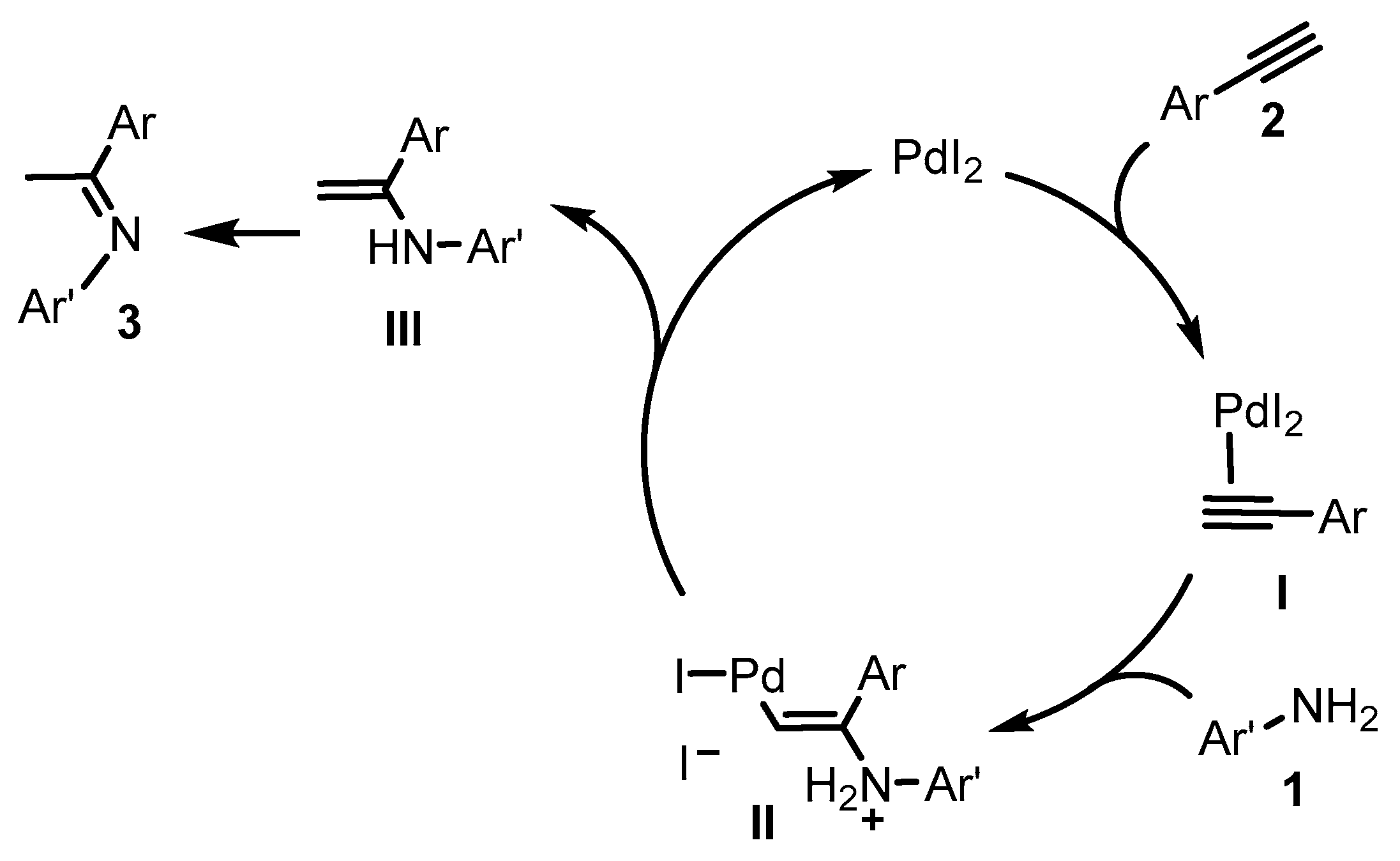
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. General Procedures
3.2.1. Hydroamination of Terminal Alkynes with Anilines
3.2.2. Reduction of Imine 3 to Secondary Amine 4
3.3. Product Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Huo, J.; He, G.; Chen, W.; Hu, X.; Deng, Q.; Chen, D. A minireview of hydroamination catalysis: Alkene and alkyne substrate selective, metal complex design. BMC Chem. 2019, 13, 89–101. [Google Scholar] [CrossRef]
- Patel, M.; Saunthwal, R.K.; Verma, A.K. Base-Mediated Hydroamination of Alkynes. Acc. Chem. Res. 2017, 50, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Arndt, M.; Gooßen, K.; Heydt, H.; Gooßen, L.J. Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev. 2015, 115, 2596–2697. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.F. Carbon–heteroatom bond formation catalysed by organometallic complexes. Nature 2008, 455, 314–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, T.E.; Hultzsch, K.C.; Yus, M.; Foubelo, F.; Tada, M. Hydroamination: Direct addition of amines to alkenes and alkynes. Chem. Rev. 2008, 108, 3795–3892. [Google Scholar] [CrossRef]
- Severin, R.; Doye, S. The catalytic hydroamination of alkynes. Chem. Soc. Rev. 2007, 36, 1407–1420. [Google Scholar] [CrossRef]
- Pohlki, F.; Doye, S. The catalytic hydroamination of alkynes. Chem. Soc. Rev. 2003, 32, 104–114. [Google Scholar] [CrossRef]
- Müller, T.E.; Beller, M. Metal-initiated amination of alkenes and alkynes. Chem. Rev. 1998, 98, 675–704. [Google Scholar] [CrossRef]
- Chen, J.; Lu, Z. Asymmetric hydrofunctionalization of minimally functionalized alkenes via earth abundant transition metal catalysis. Org. Chem. Front. 2018, 5, 260–272. [Google Scholar] [CrossRef]
- Griffin, S.E.; Pacheco, J.; Schafer, L.L. Reversible C–N Bond Formation in the Zirconium-Catalyzed Intermolecular Hydroamination of 2-Vinylpyridine. Organometallics 2019, 38, 1011–1016. [Google Scholar] [CrossRef]
- Eedugurala, N.; Hovey, M.; Ho, H.-A.; Jana, B.; Lampland, N.L.; Ellern, A.; Sadow, A.D. Cyclopentadienyl-bis(oxazoline) Magnesium and Zirconium Complexes in Aminoalkene Hydroaminations. Organometallics 2015, 34, 5566–5575. [Google Scholar] [CrossRef] [Green Version]
- Reznichenko, A.L.; Hultzsch, K.C. Early Transition Metal (Group 3–5, Lanthanides and Actinides) and Main Group Metal (Group 1, 2, and 13) Catalyzed Hydroamination. In Hydrofunctionalization; Ananikov, V.P., Tanaka, M., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; Volume 43, pp. 51–114. [Google Scholar]
- Reznichenko, A.L.; Nguyen, H.N.; Hultzsch, K.C. Asymmetric intermolecular hydroamination of unactivated alkenes with simple amines. Angew. Chem. Int. Ed. 2010, 49, 8984–8987. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Marks, T.J. Organolanthanide-Catalyzed Hydroamination. Acc. Chem. Res. 2004, 37, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Yahata, K.; Kaneko, Y.; Akai, S. Cobalt-Catalyzed Intermolecular Markovnikov Hydroamination of Nonactivated Olefins: N 2 -Selective Alkylation of Benzotriazole. Org. Lett. 2020, 22, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-H.; Lu, A.; Dong, V.M. Intermolecular Hydroamination of 1,3-Dienes To Generate Homoallylic Amines. J. Am. Chem. Soc. 2017, 139, 14049–14052. [Google Scholar] [CrossRef] [PubMed]
- Gurak, J.A.; Yang, K.S.; Liu, Z.; Engle, K.M. Directed, Regiocontrolled Hydroamination of Unactivated Alkenes via Protodepalladation. J. Am. Chem. Soc. 2016, 138, 5805–5808. [Google Scholar] [CrossRef] [PubMed]
- Sevov, C.S.; Zhou, J.; Hartwig, J.F. Iridium-Catalyzed, Intermolecular Hydroamination of Unactivated Alkenes with Indoles. J. Am. Chem. Soc. 2014, 136, 3200–3207. [Google Scholar] [CrossRef]
- Cooke, M.L.; Xu, K.; Breit, B. Enantioselective Rhodium-Catalyzed Synthesis of Branched Allylic Amines by Intermolecular Hydroamination of Terminal Allenes. Angew. Chem. Int. Ed. 2012, 51, 10876–10879. [Google Scholar] [CrossRef]
- Malhotra, D.; Mashuta, M.S.; Hammond, G.B.; Xu, B. A Highly Efficient and Broadly Applicable Cationic Gold Catalyst. Angew. Chem. Int. Ed. 2014, 53, 4456–4459. [Google Scholar] [CrossRef]
- Lavallo, V.; Wright, J.H.; Tham, F.S.; Quinlivan, S. Perhalogenated Carba-closo-dodecaborate Anions as Ligand Substituents: Applications in Gold Catalysis. Angew. Chem. Int. Ed. 2013, 52, 3172–3176. [Google Scholar] [CrossRef]
- Platon, M.; Amardeil, R.; Djakovitch, L.; Hierso, J.C. Progress in palladium-based catalytic systems for the sustainable synthesis of annulated heterocycles: A focus on indole backbones. Chem. Soc. Rev. 2012, 41, 3929–3968. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D.; Junge, K.; Beller, M. A General Catalytic Hydroamidation of 1, 3-Dienes: Atom-Efficient Synthesis of N-Allyl Heterocycles, Amides, and Sulfonamides. Angew. Chem. Int. Ed. 2014, 53, 1630–1635. [Google Scholar] [CrossRef] [PubMed]
- Lutete, L.M.; Kadota, I.; Yamamoto, Y. Palladium-catalyzed intramolecular asymmetric hydroamination of alkynes. J. Am. Chem. Soc. 2004, 126, 1622–1623. [Google Scholar] [CrossRef] [PubMed]
- Gabriele, B.; Salerno, G.; Fazio, A. General and regioselective synthesis of substituted pyrroles by Metal-Catalyzed or spontaneous cycloisomerization of (Z)-(2-En-4-ynyl) amines. J. Org. Chem. 2003, 68, 7853–7861. [Google Scholar] [CrossRef]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition metal-catalyzed addition of heteroatom−hydrogen bonds to alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef]
- Michael, F.E.; Cochran, B.M. Room temperature palladium-catalyzed intramolecular hydroamination of unactivated alkenes. J. Am. Chem. Soc. 2006, 128, 4246–4247. [Google Scholar] [CrossRef]
- Löber, O.; Kawatsura, M.; Hartwig, J.F. Palladium-catalyzed hydroamination of 1, 3-dienes: A colorimetric assay and enantioselective additions. J. Am. Chem. Soc. 2001, 123, 4366–4367. [Google Scholar] [CrossRef]
- Besson, L.; Goré, J.; Cazes, B. Synthesis of allylic amines through the palladium-catalyzed hydroamination of allenes. Tetrahedron Lett. 1995, 36, 3857–3860. [Google Scholar] [CrossRef]
- Chen, Q.; Lv, L.; Yu, M.; Shi, Y.; Li, Y.; Pang, G.; Cao, C. Simple, efficient and reusable Pd–NHC catalysts for hydroamination. RSC Adv. 2013, 3, 18359–18366. [Google Scholar] [CrossRef]
- Franco, D.; Marchenko, A.; Koidan, G.; Hurieva, A.N.; Kostyuk, A.; Biffis, A. Palladium (II) Complexes with N-Phosphanyl-N-heterocyclic Carbenes as Catalysts for Intermolecular Alkyne Hydroaminations. ACS Omega 2018, 3, 17888–17894. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, A.R.; Schmidt, J.A. Palladium (II) 3-iminophosphine complexes as intermolecular hydroamination catalysts for the formation of imines and enamines. Organometallics 2008, 27, 1259–1266. [Google Scholar] [CrossRef]
- Shimada, T.; Yamamoto, Y. Palladium-catalyzed intermolecular hydroamination of alkynes: A dramatic rate-enhancement effect of o-aminophenol. J. Am. Chem. Soc. 2002, 124, 12670–12671. [Google Scholar] [CrossRef]
- Shimada, T.; Bajracharya, G.B.; Yamamoto, Y. Aquapalladium complex: A stable and convenient catalyst for the intermolecular hydroamination of alkynes. Eur. J. Org. Chem. 2005, 2005, 59–62. [Google Scholar] [CrossRef]
- Kadota, I.; Shibuya, A.; Lutete, L.M.; Yamamoto, Y. Palladium/benzoic acid catalyzed hydroamination of alkynes. J. Org. Chem. 1999, 64, 4570–4571. [Google Scholar] [CrossRef] [PubMed]
- Casnati, A.; Perrone, A.; Mazzeo, P.P.; Bacchi, A.; Mancuso, R.; Gabriele, B.; Maggi, R.; Maestri, G.; Motti, E.; Stirling, A.; et al. Synthesis of Imidazolidin-2-ones and Imidazol-2-ones via Base-Catalyzed Intramolecular Hydroamidation of Propargylic Ureas under Ambient Conditions. J. Org. Chem. 2019, 84, 3477–3490. [Google Scholar] [CrossRef] [PubMed]
- Brunet, J.J.; Chu, N.C.; Diallo, O.; Vincendeau, S. Platinum-catalyzed intermolecular hydroamination of terminal alkynes. J. Mol. Catal. A Chem. 2005, 240, 245–248. [Google Scholar] [CrossRef]
- Gabriele, B.; Salerno, G.; Costa, M. PdI2-Catalyzed Synthesis of Heterocycles. Synlett 2004, 2004, 2468–2483. [Google Scholar] [CrossRef]
- Maitlis, P.M.; Haynes, A.; James, B.R.; Catellani, M.; Chiusoli, G.P. Iodide effects in transition metal catalyzed reactions. Dalton Trans. 2004, 21, 3409–3419. [Google Scholar] [CrossRef]
- For a recent Pd-catalyzed transformation, involving a C-N bond formation, promoted by iodide anions, see: Casnati, A.; Fontana, M.; Coruzzi, G.; Aresta, B.M.; Corriero, N.; Maggi, R.; Maestri, G.; Motti, E.; Della Ca’, N. Enhancing Reactivity and Selectivity of Aryl Bromides: A Complementary Approach to Dibenzo [b,f] azepine Derivatives. ChemCatChem 2018, 10, 4346–4352. [Google Scholar] [CrossRef]
- Liang, S.; Hammond, L.; Xu, B.; Hammond, G.B. Commercial Supported Gold Nanoparticles Catalyzed Alkyne Hydroamination and Indole Synthesis. Adv. Synth. Catal. 2016, 358, 3313–3318. [Google Scholar] [CrossRef]
- Wallach, D.R.; Stege, P.C.; Shah, J.P.; Chisholm, J.D. Brønsted acid catalyzed monoalkylation of anilines with trichloroacetimidates. J. Org. Chem. 2015, 80, 1993–2000. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Fleischer, S.; Jiao, H.; Junge, K.; Beller, M. Cooperative catalysis with iron and a chiral brønsted acid for asymmetric reductive amination of ketones. Adv. Synth. Catal. 2014, 356, 3451–3455. [Google Scholar] [CrossRef]
- Babu, N.S.; Reddy, K.M.; Prasad, P.S.; Suryanarayana, I.; Lingaiah, N. Intermolecular hydroamination of vinyl arenes using tungstophosphoric acid as a simple and efficient catalyst. Tetrahedron Lett. 2007, 48, 7642–7645. [Google Scholar] [CrossRef]
- Schroeter, F.; Lerch, S.; Kaliner, M.; Strassner, T. Cobalt-catalyzed hydroarylations and hydroaminations of alkenes in tunable aryl alkyl ionic liquids. Org. Lett. 2018, 20, 6215–6219. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Pd source (%) | Conc. (M) | T (°C) | Solvent | Conv. (%) 1a | Yieldb (%) 3a |
|---|---|---|---|---|---|---|
| 1 | Pd(OAc)2 (2) | 0.2 | 80 | 1,4-dioxane | <5 | - |
| 2 | PdCl2 (2) | 0.2 | 80 | 1,4-dioxane | 7 | 5 |
| 3 | PdI2 (2) | 0.2 | 80 | 1,4-dioxane | 91 | 73 |
| 4 | PdI2 (2) + KI (20%) | 0.2 | 80 | 1,4-dioxane | 82 | 64 |
| 5 | K2PdI4 (2) | 0.2 | 80 | 1,4-dioxane | 81 | 62 |
| 6 | PdI2 (2) | 0.4 | 80 | 1,4-dioxane | 99 | 81 |
| 7 | PdI2 (2) | 0.8 | 80 | 1,4-dioxane | 95 | 74 |
| 8 | PdI2 (2) | 0.4 | 100 | 1,4-dioxane | 99 | 77 |
| 9 | PdI2 (1) | 0.4 | 80 | 1,4-dioxane | 84 | 63 |
| 10 | PdI2 (0.5) | 0.4 | 80 | 1,4-dioxane | 69 | 47 |
| 11 | PdI2 (2) | 0.4 | 80 | DMF | <5 | 0 |
| 12 | PdI2 (2) | 0.4 | 80 | MeCN | 38 | 27 |
| 13 | PdI2 (2) | 0.4 | 80 | toluene | 47 | 36 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casnati, A.; Voronov, A.; Ferrari, D.G.; Mancuso, R.; Gabriele, B.; Motti, E.; Della Ca’, N. PdI2 as a Simple and Efficient Catalyst for the Hydroamination of Arylacetylenes with Anilines. Catalysts 2020, 10, 176. https://doi.org/10.3390/catal10020176
Casnati A, Voronov A, Ferrari DG, Mancuso R, Gabriele B, Motti E, Della Ca’ N. PdI2 as a Simple and Efficient Catalyst for the Hydroamination of Arylacetylenes with Anilines. Catalysts. 2020; 10(2):176. https://doi.org/10.3390/catal10020176
Chicago/Turabian StyleCasnati, Alessandra, Aleksandr Voronov, Damiano Giuseppe Ferrari, Raffaella Mancuso, Bartolo Gabriele, Elena Motti, and Nicola Della Ca’. 2020. "PdI2 as a Simple and Efficient Catalyst for the Hydroamination of Arylacetylenes with Anilines" Catalysts 10, no. 2: 176. https://doi.org/10.3390/catal10020176