Role of Cobalt(III) Cationic Complexes in the Self-Assembling Process of a Water Soluble Porphyrin

 , , and
, , and
Abstract
:1. Introduction
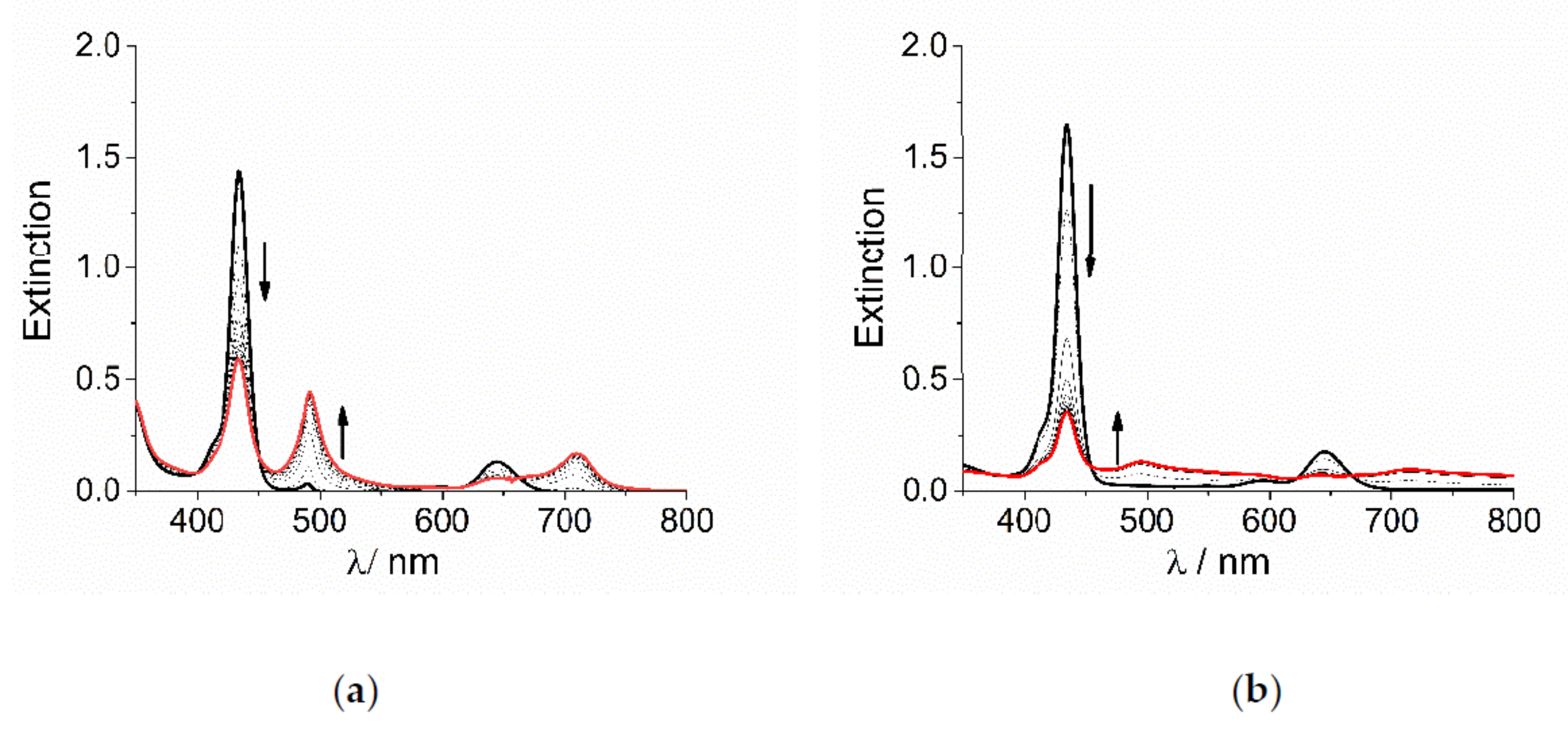
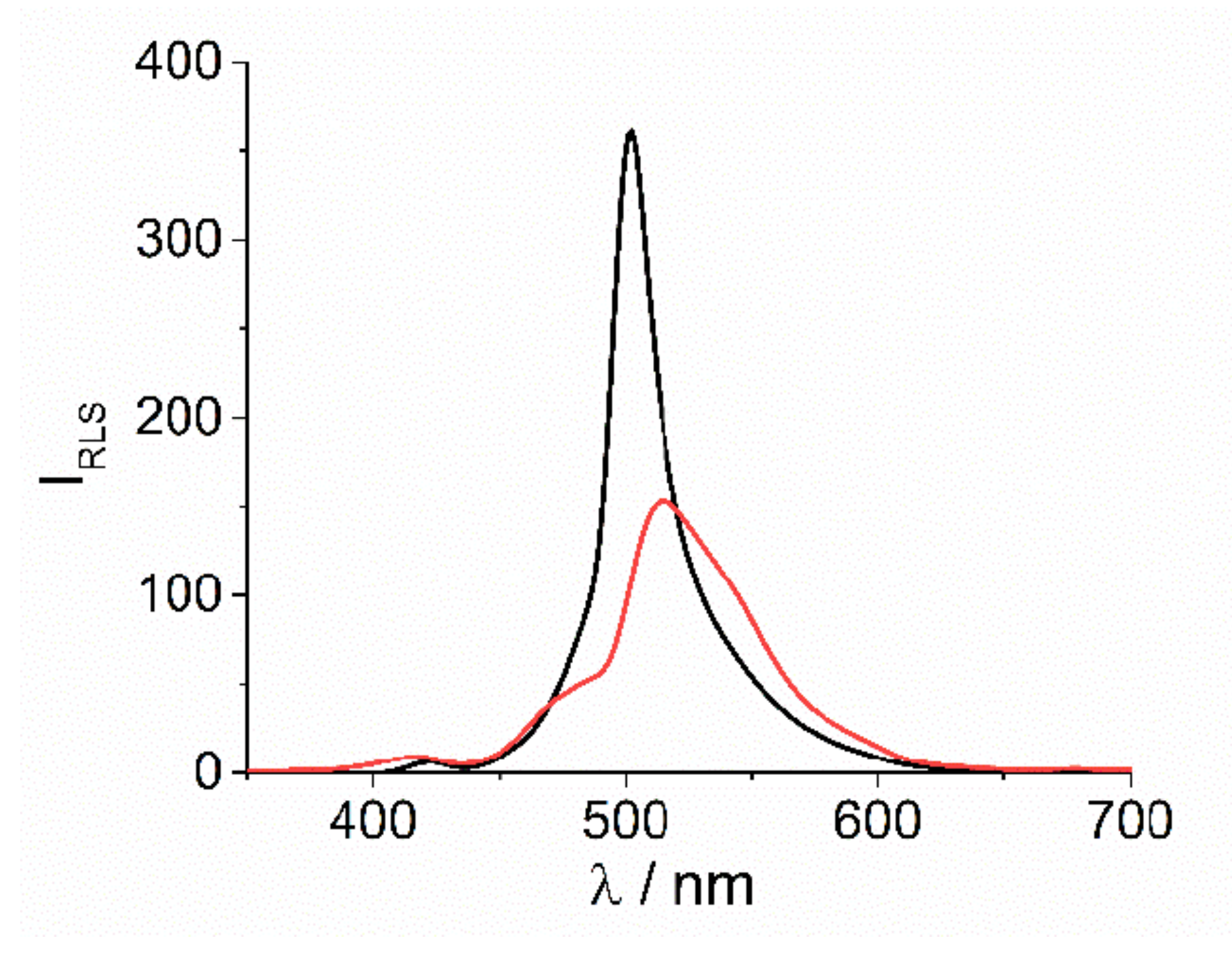
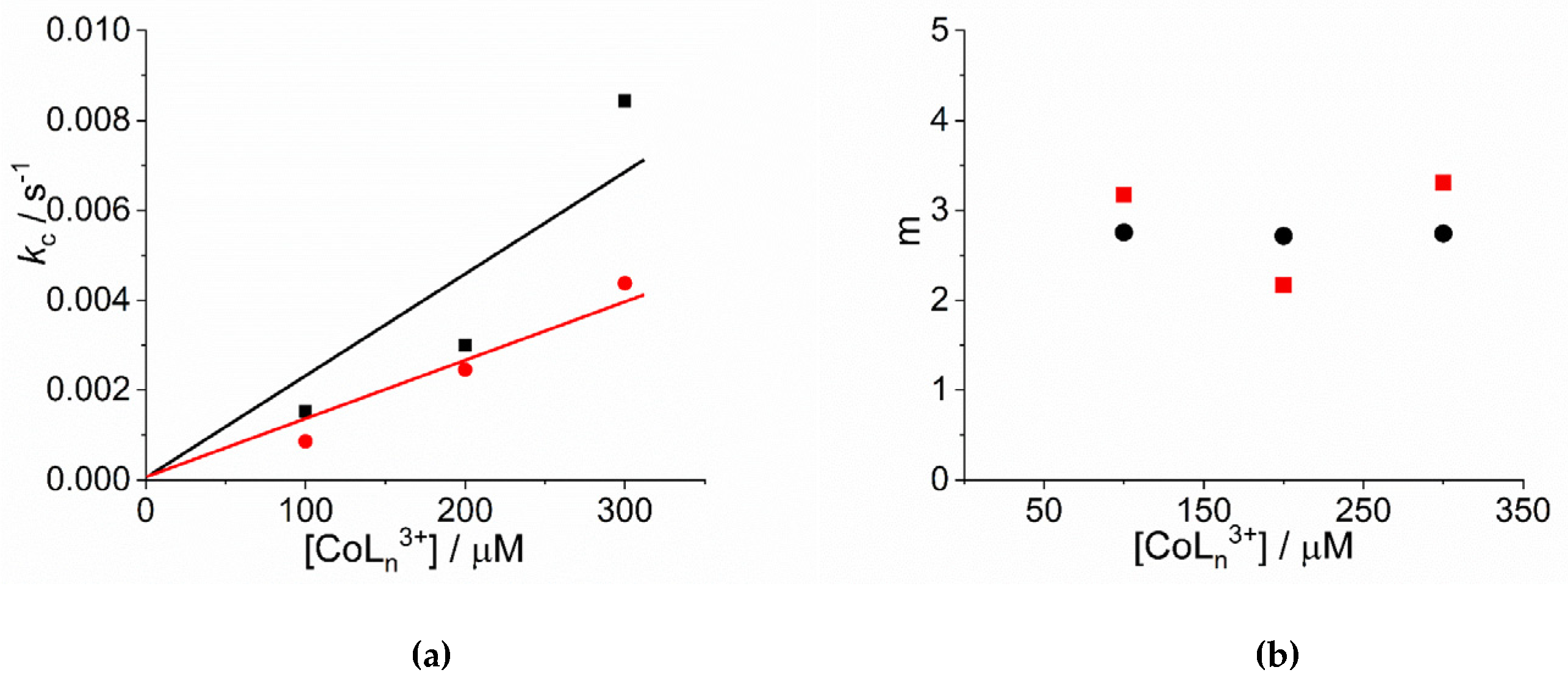
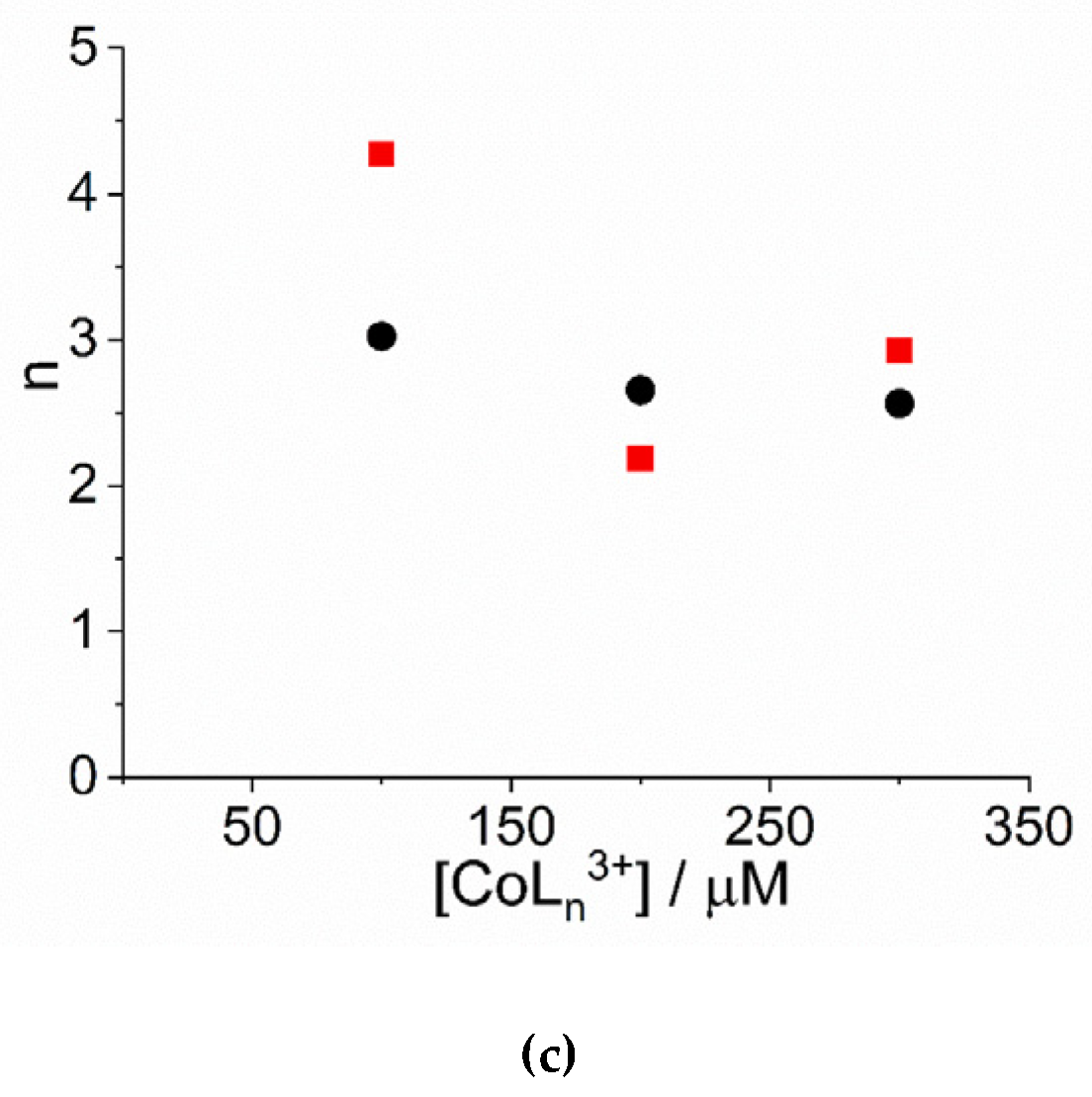
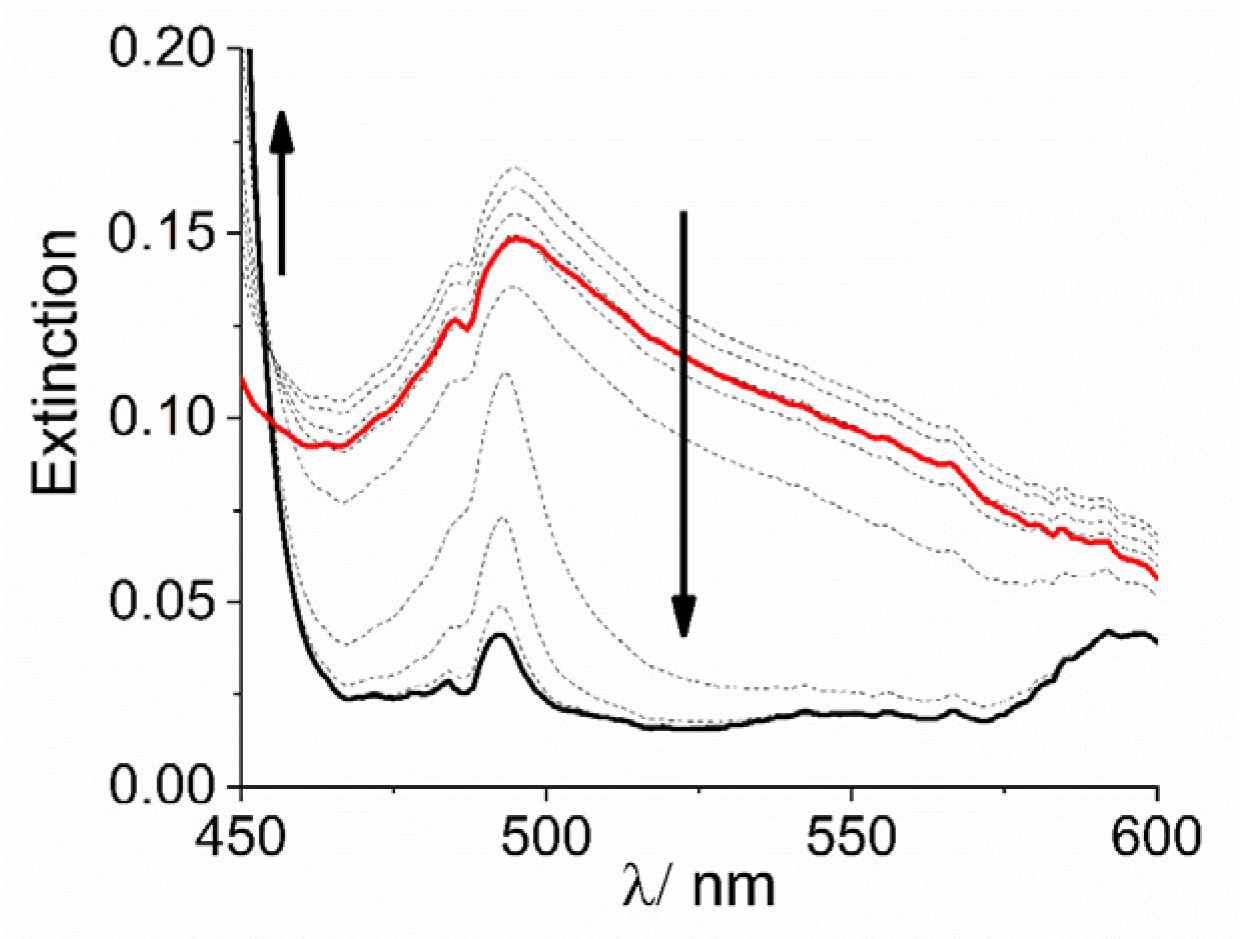
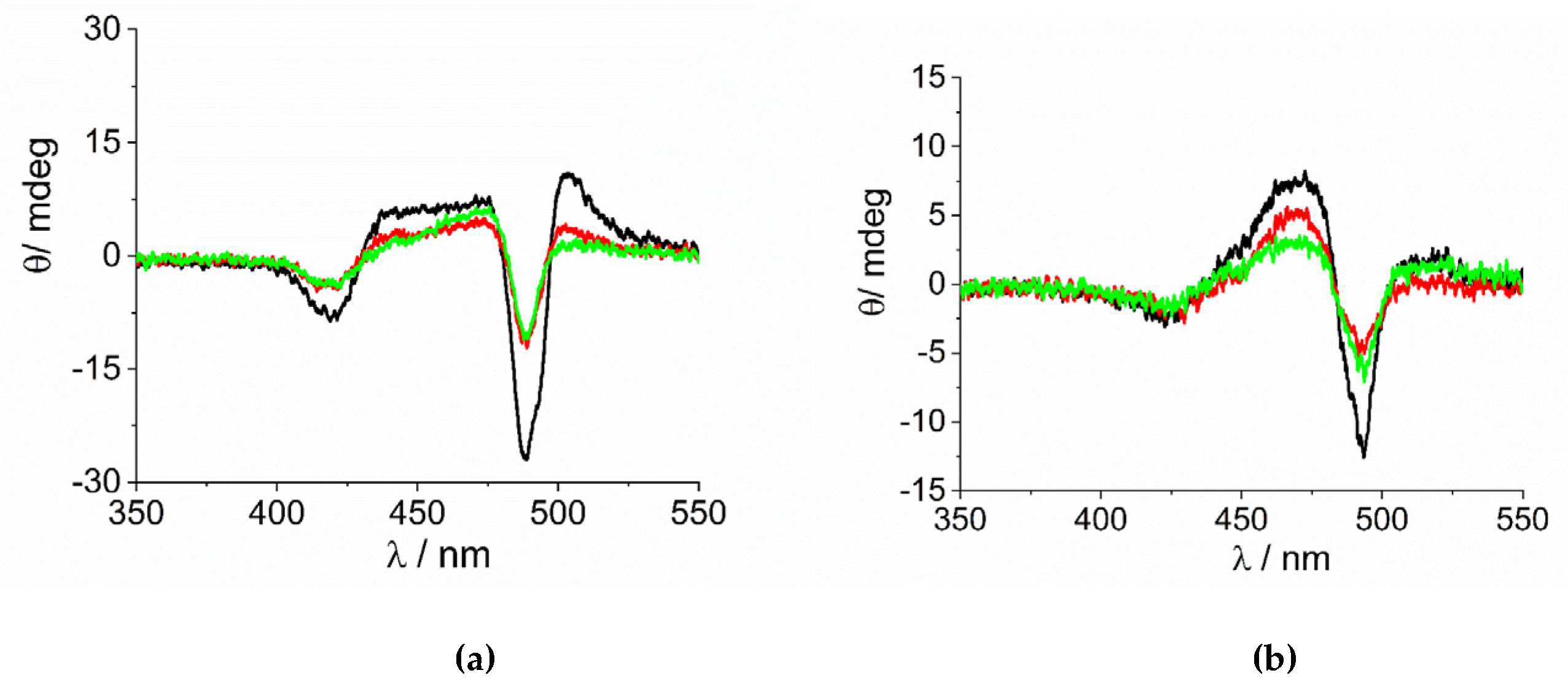
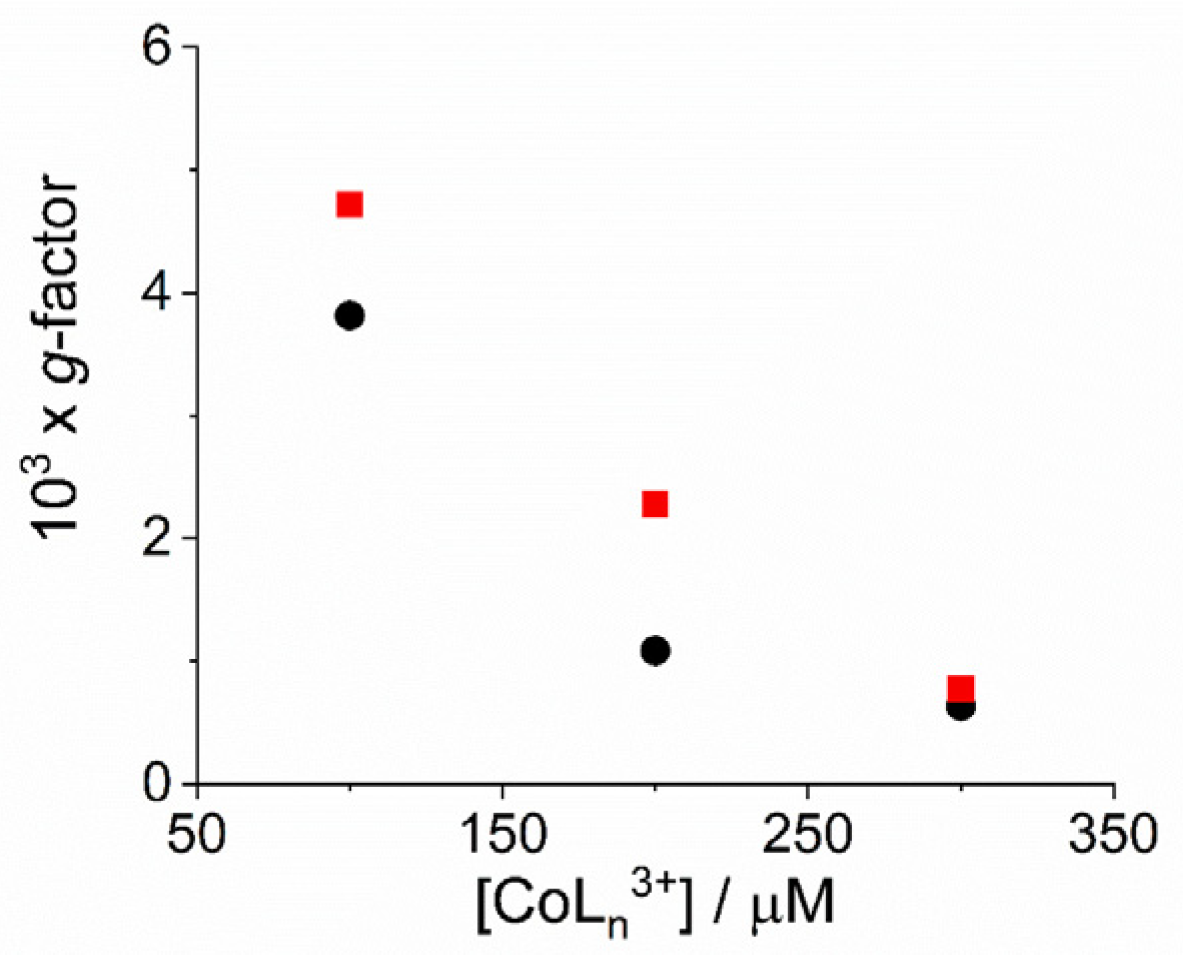
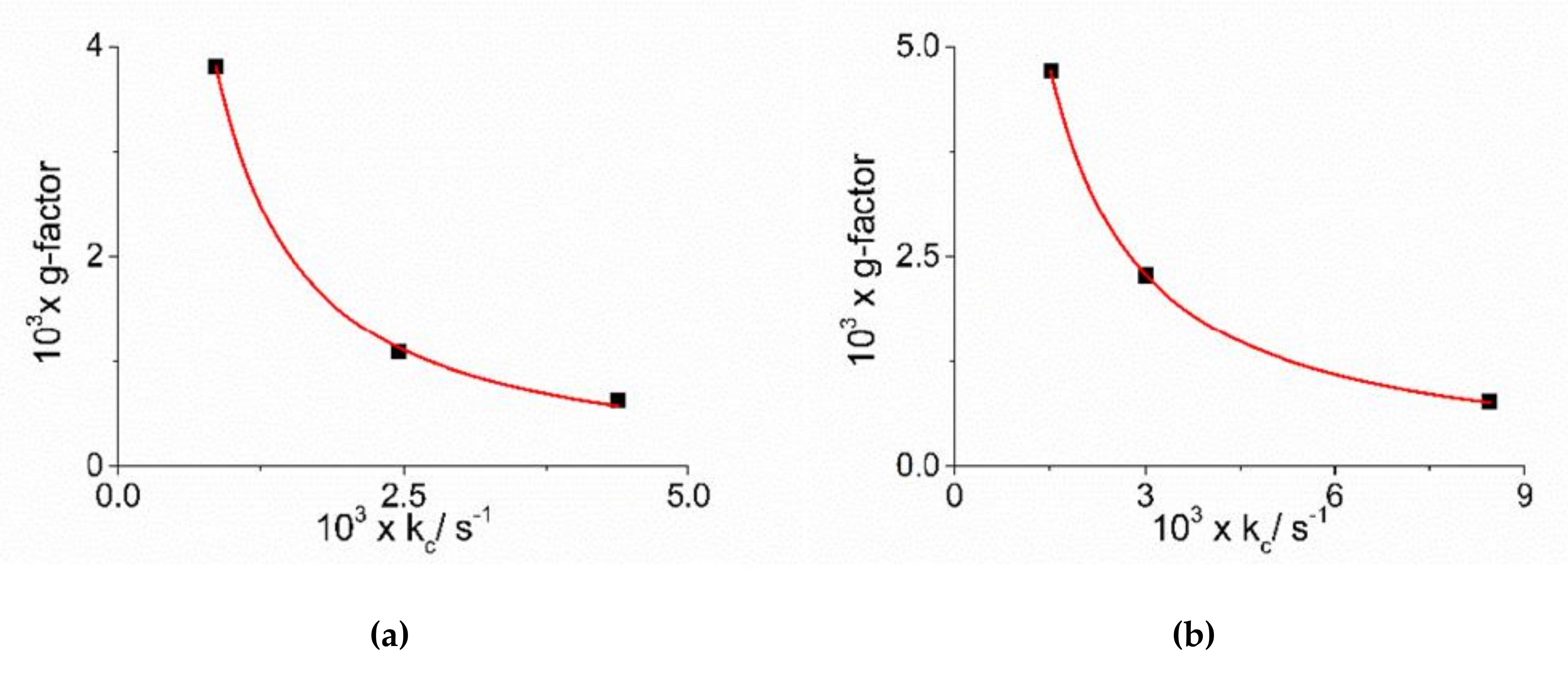
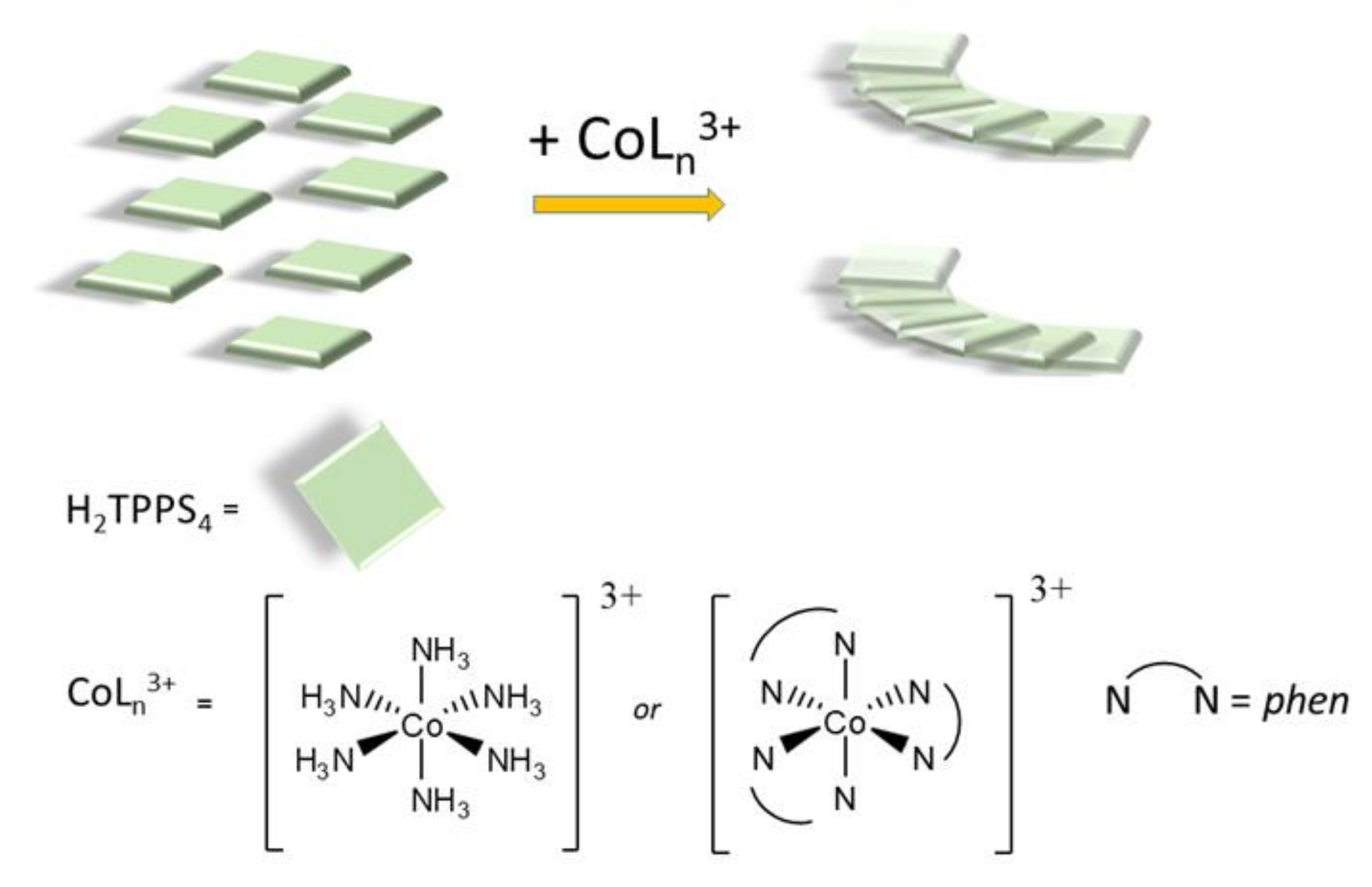
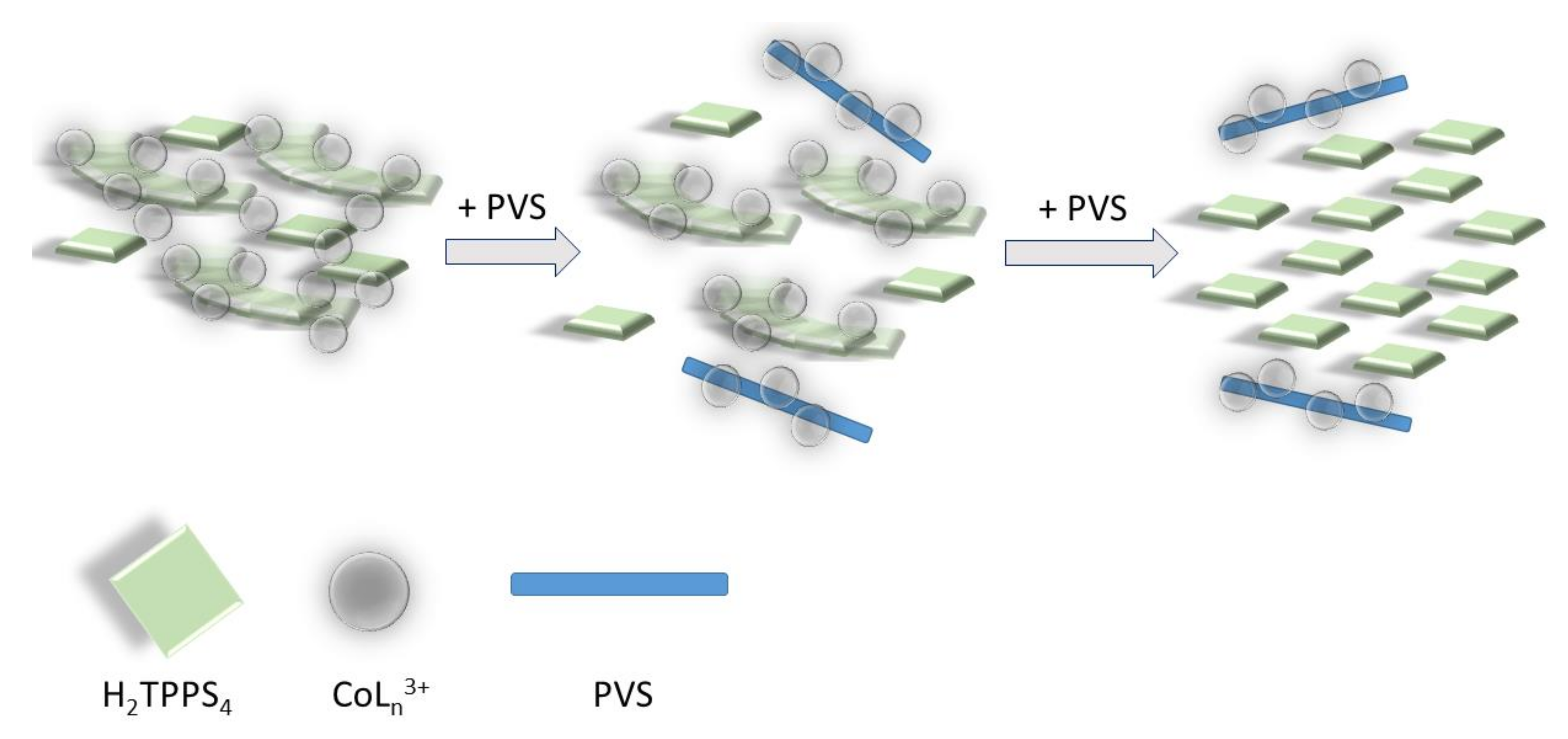
2. Results and Discussion
3. Materials and Methods
3.1. Materials
3.2. Methods
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- White, W.I. 7—Aggregation of Porphyrins and Metalloporphyrins. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; pp. 303–339. [Google Scholar] [CrossRef]
- Magna, G.; Monti, D.; Di Natale, C.; Paolesse, R.; Stefanelli, M. The Assembly of Porphyrin Systems in Well-Defined Nanostructures: An Update. Molecules 2019, 24, 4307. [Google Scholar] [CrossRef] [Green Version]
- Stefanelli, M.; Mandoj, F.; Magna, G.; Lettieri, R.; Venanzi, M.; Paolesse, R.; Monti, D. The Self-Aggregation of Porphyrins with Multiple Chiral Centers in Organic/Aqueous Media: The Case of Sugar- and Steroid-Porphyrin Conjugates. Molecules 2020, 25, 4544. [Google Scholar] [CrossRef]
- Monti, D.; Nardis, S.; Stefanelli, M.; Paolesse, R.; Di Natale, C.; D’Amico, A. Porphyrin-Based Nanostructures for Sensing Applications. J. Sens. 2009, 2009. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.B.; Xu, J.L.; Li, Y.J.; Li, Y.L. Aggregate Nanostructures of Organic Molecular Materials. Acc. Chem. Res. 2010, 43, 1496–1508. [Google Scholar] [CrossRef] [PubMed]
- Bajjou, O.; Mongwaketsi, P.N.; Khenfouch, M.; Bakour, A.; Baitoul, M.; Maaza, M.; Venturini, J.W. Photoluminescence Quenching and Structure of Nanocomposite Based on Graphene Oxide Layers Decorated with Nanostructured Porphyrin. Nanomater. Nanotechnol. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Mondal, B.; Bera, R.; Nayak, S.K.; Patra, A. Graphene induced porphyrin nano-aggregates for efficient electron transfer and photocurrent generation. J. Mater. Chem. C 2016, 4, 6027–6036. [Google Scholar] [CrossRef]
- Lu, J.R.; Li, Z.H.; An, W.J.; Liu, L.; Cui, W.Q. Tuning the Supramolecular Structures of Metal-Free Porphyrin via Surfactant Assisted Self-Assembly to Enhance Photocatalytic Performance. Nanomaterials 2019, 9, 1321. [Google Scholar] [CrossRef] [Green Version]
- Shinokubo, H.; Osuka, A. Marriage of porphyrin chemistry with metal-catalysed reactions. Chem. Commun. 2009, 1011–1021. [Google Scholar] [CrossRef]
- Mohnani, S.; Bonifazi, D. Supramolecular architectures of porphyrins on surfaces: The structural evolution from 1D to 2D to 3D to devices. Coord. Chem. Rev. 2010, 254, 2342–2362. [Google Scholar] [CrossRef]
- Kasha, M.; Rawls, H.R.; Ashraf El-Bayoumi, M. The exciton model in molecular spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. [Google Scholar] [CrossRef] [Green Version]
- Pasternack, R.F.; Collings, P.J. Resonance Light-Scattering—A New Technique for Studying Chromophore Aggregation. Science 1995, 269, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Collings, P.J.; Gibbs, E.J.; Starr, T.E.; Vafek, O.; Yee, C.; Pomerance, L.A.; Pasternack, R.F. Resonance light scattering and its application in determining the size, shape, and aggregation number for supramolecular assemblies of chromophores. J. Phys. Chem. B 1999, 103, 8474–8481. [Google Scholar] [CrossRef]
- Villari, V.; Fazio, B.; Micali, N.; De Luca, G.; Corsaro, C.; Romeo, A.; Scolaro, L.M.; Castriciano, M.A.; Mazzaglia, A. Light scattering enhancement in porphyrin nanocomposites. In Complex Materials in Physics and Biology; Mallamace, F., Stanley, H.E., Eds.; IOS: Amsterdam, The Netherlands, 2012; Volume 176, pp. 335–340. [Google Scholar]
- Villari, V.; Fazio, B.; De Luca, G.; Trapani, M.; Romeo, A.; Scolaro, L.M.; Castriciano, M.A.; Mazzaglia, A.; Micali, N. Scattering enhancement in colloidal metal-organic composite aggregates. Colloids Surf. A Physicochem. Eng. Asp. 2012, 413, 13–16. [Google Scholar] [CrossRef]
- Scolaro, L.M.; Romeo, A.; Castriciano, M.A.; Micali, N. Unusual optical properties of porphyrin fractal J-aggregates. Chem. Commun. 2005, 3018–3020. [Google Scholar] [CrossRef] [PubMed]
- Micali, N.; Villari, V.; Scolaro, L.M.; Romeo, A.; Castriciano, M.A. Light scattering enhancement in an aqueous solution of spermine-induced fractal J-aggregate composite. Phys. Rev. E 2005, 72. [Google Scholar] [CrossRef]
- Micali, N.; Mallamace, F.; Castriciano, M.; Romeo, A.; Scolaro, L.M. Separation of scattering and absorption contributions in UV/visible spectra of resonant systems. Anal. Chem. 2001, 73, 4958–4963. [Google Scholar] [CrossRef] [PubMed]
- Castriciano, M.A.; Donato, M.G.; Villari, V.; Micali, N.; Romeo, A.; Scolaro, L.M. Surfactant-like Behavior of Short-Chain Alcohols in Porphyrin Aggregation. J. Phys. Chem. B 2009, 113, 11173–11178. [Google Scholar] [CrossRef]
- Villari, V.; Mazzaglia, A.; Castriciano, M.A.; Luca, G.d.; Romeo, A.; Scolaro, L.M.; Micali, N. Optical enhancement and structural properties of a hybrid organic-inorganic ternary nanocomposite. J. Phys. Chem. C 2011, 115, 5435–5439. [Google Scholar] [CrossRef]
- Micali, N.; Villari, V.; Romeo, A.; Castriciano, M.A.; Scolaro, L.M. Evidence of the early stage of porphyrin aggregation by enhanced Raman scattering and fluorescence spectroscopy. Phys. Rev. E 2007, 76, 011404. [Google Scholar] [CrossRef]
- Trapani, M.; Castriciano, M.A.; Romeo, A.; De Luca, G.; Machado, N.; Howes, B.D.; Smulevich, G.; Scolaro, L.M. Nanohybrid Assemblies of Porphyrin and Au-10 Cluster Nanoparticles. Nanomaterials 2019, 9, 1026. [Google Scholar] [CrossRef] [Green Version]
- Akins, D.L.; Zhu, H.R.; Guo, C. Absorption and Raman Scattering by Aggregated meso-Tetrakis(p-sulfonatophenyl)porphine. J. Phys. Chem. 1994, 98, 3612–3618. [Google Scholar] [CrossRef]
- Rich, C.C.; McHale, J.L. Resonance Raman Spectra of Individual Excitonically Coupled Chromophore Aggregates. J. Phys. Chem. C 2013, 117, 10856–10865. [Google Scholar] [CrossRef]
- Friesen, B.A.; Rich, C.C.; Mazur, U.; McHale, J.L. Resonance Raman Spectroscopy of Helical Porphyrin Nanotubes. J. Phys. Chem. C 2010, 114, 16357–16366. [Google Scholar] [CrossRef]
- Collini, E.; Ferrante, C.; Bozio, R. Strong Enhancement of the Two-Photon Absorption of Tetrakis(4-sulfonatophenyl)porphyrin Diacid in Water upon Aggregation. J. Phys. Chem. B 2005, 109, 2–5. [Google Scholar] [CrossRef]
- Collini, E.; Ferrante, C.; Bozio, R.; Lodi, A.; Ponterini, G. Large third-order nonlinear optical response of porphyrin J-aggregates oriented in self-assembled thin films. J. Mater. Chem. 2006, 16, 1573–1578. [Google Scholar] [CrossRef]
- Collini, E.; Ferrante, C.; Bozio, R. Influence of excitonic interactions on the transient absorption and two-photon absorption spectra of porphyrin J-aggregates in the NIR region. J. Phys. Chem. C 2007, 111, 18636–18645. [Google Scholar] [CrossRef]
- Bolzonello, L.; Fassioli, F.; Collini, E. Correlated Fluctuations and Intraband Dynamics of J-Aggregates Revealed by Combination of 2DES Schemes. J. Phys. Chem. Lett. 2016, 7, 4996–5001. [Google Scholar] [CrossRef]
- Bolzonello, L.; Albertini, M.; Collini, E.; Di Valentin, M. Delocalized triplet state in porphyrin J-aggregates revealed by EPR spectroscopy. Phys. Chem. Chem. Phys. 2017, 19, 27173–27177. [Google Scholar] [CrossRef]
- Volpato, A.; Zerbetto, M.; Bolzonello, L.; Meneghin, E.; Fresch, B.; Benelli, T.; Giorgini, L.; Collini, E. Effect of Different Conformational Distributions on the Ultrafast Coherence Dynamics in Porphyrin-Based Polymers. J. Phys. Chem. C 2019, 123, 10212–10224. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Romeo, A.; Zagami, R.; Micali, N.; Scolaro, L.M. Kinetic effects of tartaric acid on the growth of chiral J-aggregates of tetrakis(4-sulfonatophenyl)porphyrin. Chem. Commun. 2012, 48, 4872–4874. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Romeo, A.; De Luca, G.; Villari, V.; Scolaro, L.M.; Micali, N. Scaling the Chirality in Porphyrin J-Nanoaggregates. J. Am. Chem. Soc. 2011, 133, 765–767. [Google Scholar] [CrossRef] [PubMed]
- Purrello, R.; Scolaro, L.M.; Bellacchio, E.; Gurrieri, S.; Romeo, A. Chiral H- and J-Type Aggregates of meso-Tetrakis(4-sulfonatophenyl)porphine on α-Helical Polyglutamic Acid Induced by Cationic Porphyrins. Inorg. Chem. 1998, 37, 3647–3648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lu, Q.; Liu, M. Fabrication of Chiral Langmuir-Schaefer Films from Achiral TPPS and Amphiphiles through the Adsorption at the Air/Water Interface. J. Phys. Chem. B 2003, 107, 2565–2569. [Google Scholar] [CrossRef]
- Zhang, L.; Yuan, J.; Liu, M. Supramolecular Chirality of Achiral TPPS Complexed with Chiral Molecular Films. J. Phys. Chem. B 2003, 107, 12768–12773. [Google Scholar] [CrossRef]
- Jiang, S.; Liu, M. Aggregation and Induced Chirality of an Anionic meso-Tetraphenylsulfonato Porphyrin (TPPS) on a Layer-by-Layer Assembled DNA/PAH Matrix. J. Phys. Chem. B 2004, 108, 2880–2884. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, M.H. Supramolecular Chirality and Chiral Inversion of Tetraphenylsulfonato Porphyrin Assemblies on Optically Active Polylysine. J. Phys. Chem. B 2009, 113, 14015–14020. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, X.; Li, Y.; Ma, R.; An, Y.; Shi, L. Chiral Micelles of Achiral TPPS and Diblock Copolymer Induced by Amino Acids. Macromolecules 2009, 42, 6253–6260. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Escudero, C.; Acosta-Reyes, F.; Casas, M.T.; Altoe, V.; Aloni, S.; Oncins, G.; Sorrenti, A.; Crusats, J.; Campos, J.L.; et al. Structure vs. Properties—chirality, optics and shapes—in amphiphilic porphyrin J-aggregates. J. Mater. Chem. C 2013, 1, 3337–3346. [Google Scholar] [CrossRef] [Green Version]
- Randazzo, R.; Gaeta, M.; Gangemi, C.M.A.; Fragalà, M.E.; Purrello, R.; D’Urso, A. Chiral Recognition of L- and D- Amino Acid by Porphyrin Supramolecular Aggregates. Molecules 2018, 24, 84. [Google Scholar] [CrossRef] [Green Version]
- Trapani, M.; Mazzaglia, A.; Piperno, A.; Cordaro, A.; Zagami, R.; Castriciano, M.A.; Romeo, A.; Monsù Scolaro, L. Novel Nanohybrids Based on Supramolecular Assemblies of Meso-tetrakis-(4-sulfonatophenyl) Porphyrin J-aggregates and Amine-Functionalized Carbon Nanotubes. Nanomaterials 2020, 10, 669. [Google Scholar] [CrossRef] [Green Version]
- El-Hachemi, Z.; Balaban, T.S.; Campos, J.L.; Cespedes, S.; Crusats, J.; Escudero, C.; Kamma-Lorger, C.S.; Llorens, J.; Malfois, M.; Mitchell, G.R.; et al. Effect of Hydrodynamic Forces on meso-(4-Sulfonatophenyl)-Substituted Porphyrin J-Aggregate Nanoparticles: Elasticity, Plasticity and Breaking. Chem. A Eur. J. 2016, 22, 9740–9749. [Google Scholar] [CrossRef] [PubMed]
- Escudero, C.; Crusat, J.; Diez-Perez, I.; El-Hachemi, Z.; Ribo, J.M. Folding and hydrodynamic forces in J-aggregates of 5-phenyl-10,15,20-tris-(4-sulfo-phenyl)porphyrin. Angew. Chem. Int. Ed. 2006, 45, 8032–8035. [Google Scholar] [CrossRef] [PubMed]
- Ribo, J.M.; Crusats, J.; Sagues, F.; Claret, J.; Rubires, R. Chiral sign induction by vortices during the formation of mesophases in stirred solutions. Science 2001, 292, 2063. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, A.; Randazzo, R.; Lo Faro, L.; Purrello, R. Vortexes and Nanoscale Chirality. Angew. Chem. Int. Edn Engl. 2010, 49, 108–112. [Google Scholar] [CrossRef]
- Crusats, J.; El-Hachemi, Z.; Ribo, J.M. Hydrodynamic effects on chiral induction. Chem. Soc. Rev. 2010, 39, 569. [Google Scholar] [CrossRef]
- Micali, N.; Engelkamp, H.; van Rhee, P.G.; Christianen, P.C.M.; Scolaro, L.M.; Maan, J.C. Selection of supramolecular chirality by application of rotational and magnetic forces. Nat. Chem. 2012, 4, 201–207. [Google Scholar] [CrossRef]
- Arteaga, O.; Canillas, A.; Purrello, R.; Ribo, J.M. Evidence of induced chirality in stirred solutions of supramolecular nanofibers. Opt. Lett. 2009, 34, 2177–2179. [Google Scholar] [CrossRef]
- Sun, J.S.; Li, Y.K.; Yan, F.S.; Liu, C.; Sang, Y.T.; Tian, F.; Feng, Q.; Duan, P.F.; Zhang, L.; Shi, X.H.; et al. Control over the emerging chirality in supramolecular gels and solutions by chiral microvortices in milliseconds. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Short, J.M.; Berriman, J.A.; Kübel, C.; El-Hachemi, Z.; Naubron, J.-V.; Balaban, T.S. Electron Cryo-Microscopy of TPPS4 2HCl Tubes Reveals a Helical Organisation Explaining the Origin of their Chirality. ChemPhysChem 2013, 14, 3209–3214. [Google Scholar] [CrossRef] [Green Version]
- Balaban, T.S.; Bhise, A.D.; Fischer, M.; Linke-Schaetzel, M.; Roussel, C.; Vanthuyne, N. Controlling Chirality and Optical Properties of Artificial Antenna Systems with Self-Assembling Porphyrins. Angew. Chem. Int. Edn Engl. 2003, 42, 2140–2144. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Huber, P.R.; Boyd, P.; Engasser, G.; Francesconi, L.; Gibbs, E.; Fasella, P.; Cerio Venturo, G.; Hinds Lde, C. On the Aggregation of Meso-Substituted Water-S’oluble Porphyrins. J. Am. Chem. Soc. 1972, 94, 4511–4517. [Google Scholar] [CrossRef] [PubMed]
- Ohno, O.; Kaizu, Y.; Kobayashi, H. J-Aggregate Formation of a Water-Soluble Porphyrin in Acidic Aqueous-Media. J. Chem. Phys. 1993, 99, 4128–4139. [Google Scholar] [CrossRef]
- Maiti, N.C.; Mazumdar, S.; Periasamy, N. J- and H-aggregates of porphyrin-surfactant complexes: Time-resolved fluorescence and other spectroscopic studies. J. Phys. Chem. B 1998, 102, 1528–1538. [Google Scholar] [CrossRef]
- Akins, D.L.; Zhu, H.R.; Guo, C. Aggregation of tetraaryl-substituted porphyrins in homogeneous solution. J. Phys. Chem. 1996, 100, 5420–5425. [Google Scholar] [CrossRef]
- Ribo, J.M.; Crusats, J.; Farrera, J.A.; Valero, M.L. Aggregation in Water Solutions of Tetrasodium Diprotonated Meso-Tetrakis(4-Sulfonatophenyl)Porphyrin. J. Chem. Soc., Chem. Commun. 1994, 6, 681–682. [Google Scholar] [CrossRef]
- Maiti, N.C.; Ravikanth, M.; Mazumdar, S.; Periasamy, N. Fluorescence Dynamics of Noncovalently Linked Porphyrin Dimers, and Aggregates. J. Phys. Chem. 1995, 99, 17192–17197. [Google Scholar] [CrossRef]
- Micali, N.; Romeo, A.; Lauceri, R.; Purrello, R.; Mallamace, F.; Scolaro, L.M. Fractal structures in homo- and heteroaggregated water soluble porphyrins. J. Phys. Chem. B 2000, 104, 9416–9420. [Google Scholar] [CrossRef]
- Micali, N.; Mallamace, F.; Romeo, A.; Purrello, R.; Scolaro, L.M. Mesoscopic structure of meso-tetrakis(4-sulfonatophenyl)porphine J-aggregates. J. Phys. Chem. B 2000, 104, 5897–5904. [Google Scholar] [CrossRef]
- Micali, N.; Villari, V.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M. From fractal to nanorod porphyrin J-aggregates. Concentration-induced tuning of the aggregate size. J. Phys. Chem. B 2006, 110, 8289–8295. [Google Scholar] [CrossRef]
- Zagami, R.; Romeo, A.; Castriciano, M.A.; Monsù Scolaro, L. Inverse Kinetic and Equilibrium Isotope Effects on Self-Assembly and Supramolecular Chirality of Porphyrin J-Aggregates. Chem. A Eur. J. 2017, 23, 70–74. [Google Scholar] [CrossRef]
- Occhiuto, I.G.; Zagami, R.; Trapani, M.; Bolzonello, L.; Romeo, A.; Castriciano, M.A.; Collini, E.; Monsu Scolaro, L. The role of counter-anions in the kinetics and chirality of porphyrin J-aggregates. Chem. Commun. 2016, 52, 11520–11523. [Google Scholar] [CrossRef] [PubMed]
- Friesen, B.A.; Nishida, K.R.A.; McHale, J.L.; Mazur, U. New Nanoscale Insights into the Internal Structure of Tetrakis(4-sulfonatophenyl) Porphyrin Nanorods. J. Phys. Chem. C 2009, 113, 1709–1718. [Google Scholar] [CrossRef]
- Occhiuto, I.G.; Castriciano, M.A.; Trapani, M.; Zagami, R.; Romeo, A.; Pasternack, R.F.; Monsù Scolaro, L. Controlling J-Aggregates Formation and Chirality Induction through Demetallation of a Zinc(II) Water Soluble Porphyrin. Int. J. Mol. Sci. 2020, 21, 4001. [Google Scholar] [CrossRef] [PubMed]
- Castriciano, M.A.; Trapani, M.; Romeo, A.; Depalo, N.; Rizzi, F.; Fanizza, E.; Patanè, S.; Monsù Scolaro, L. Influence of Magnetic Micelles on Assembly and Deposition of Porphyrin J-Aggregates. Nanomaterials 2020, 10, 187. [Google Scholar] [CrossRef] [Green Version]
- Zagami, R.; Castriciano, M.A.; Romeo, A.; Trapani, M.; Pedicini, R.; Scolaro, L.M. Tuning supramolecular chirality in nano and mesoscopic porphyrin J-aggregates. Dye. Pigment. 2017, 142, 255–261. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Leone, N.; Cardiano, P.; Manickam, S.; Scolaro, L.M.; Lo Schiavo, S. A new supramolecular polyhedral oligomeric silsesquioxanes (POSS)-porphyrin nanohybrid: Synthesis and spectroscopic characterization. J. Mater. Chem. C 2013, 1, 4746–4753. [Google Scholar] [CrossRef]
- Lauceri, R.; Gurrieri, S.; Bellacchio, E.; Contino, A.; Scolaro, L.M.; Romeo, A.; Toscano, A.; Purrello, R. J-type aggregates of the anionic meso-tetrakis(4-sulfonatophenyl)porphine induced by “hindered” cationic porphyrins. Supramol. Chem. 2000, 12, 193–202. [Google Scholar] [CrossRef]
- Randazzo, R.; Mammana, A.; D’Urso, A.; Lauceri, R.; Purrello, R. Reversible “Chiral Memory” in Ruthenium Tris(phenanthroline)-Anionic Porphyrin Complexes. Angew. Chem. Int. Ed. 2008, 47, 9879–9882. [Google Scholar] [CrossRef]
- Wang, J.; Liu, C.; Ding, D.; Zeng, L.; Cao, Q.; Zhang, H.; Zhao, H.; Li, X.; Xiang, K.; He, Y.; et al. Aggregation of an anionic porphyrin with chiral metal complexes and the competitive binding influences of a surfactant and a polyelectrolyte. New J. Chem. 2011, 35, 1424–1432. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ding, D.; Zeng, L.; Cao, J.; He, Y.; Zhang, H. Transformation, memorization and amplification of chirality in cationic Co(III) complex–porphyrin aggregates. New J. Chem. 2010, 34, 1394–1400. [Google Scholar] [CrossRef] [Green Version]
- Castriciano, M.A.; Romeo, A.; Scolaro, L.M. Aggregation of meso-tetrakis(4-sulfonatophenyl)porphyrin on polyethyleneimine in aqueous solutions and on a glass surface. J. Porphyr. Phthalocyanines 2002, 6, 431–438. [Google Scholar] [CrossRef]
- Trapani, M.; Occhiuto, I.G.; Zagami, R.; De Luca, G.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M.; Pasternack, R.F. Mechanism for Copper(II)-Mediated Disaggregation of a Porphyrin J-Aggregate. ACS Omega 2018, 3, 18843–18848. [Google Scholar] [CrossRef] [PubMed]
- Occhiuto, I.; De Luca, G.; Trapani, M.; Scolaro, L.M.; Pasternack, R.F. Peripheral Stepwise Degradation of a Porphyrin J-Aggregate. Inorg. Chem. 2012, 51, 10074–10076. [Google Scholar] [CrossRef] [PubMed]
- Romeo, A.; Castriciano, M.A.; Scolaro, L.M. Spectroscopic and kinetic investigations on porphyrin J-aggregates induced by polyamines. J. Porphyr. Phthalocyanines 2010, 14, 713–721. [Google Scholar] [CrossRef]
- Romeo, A.; Castriciano, M.A.; Occhiuto, I.; Zagami, R.; Pasternack, R.F.; Scolaro, L.M. Kinetic Control of Chirality in Porphyrin J-Aggregates. J. Am. Chem. Soc. 2014, 136, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, K. Photochemistry of Polypyridine and Porphyrin Complexes; Academic Press: London, UK, 1992; p. 428. [Google Scholar]
- Castriciano, M.; Romeo, A.; Villari, V.; Micali, N.; Scolaro, L.M. Structural rearrangements in 5,10,15,20-tetrakis(4-sulfonatophenyl)porphyrin J-aggregates under strongly acidic conditions. J. Phys. Chem. B 2003, 107, 8765–8771. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Fleming, C.; Herring, S.; Collings, P.J.; dePaula, J.; DeCastro, G.; Gibbs, E.J. Aggregation kinetics of extended porphyrin and cyanine dye assemblies. Biophys. J. 2000, 79, 550–560. [Google Scholar] [CrossRef] [Green Version]
- Castriciano, M.A.; Samperi, M.; Camiolo, S.; Romeo, A.; Scolaro, L.M. Unusual Stepwise Protonation and J-Aggregation of meso-Tetrakis(N-methylpyridinium-4-yl)porphine on Binding Poly(sodium vinylsulfonate). Chem. A Eur. J. 2013, 19, 12161–12168. [Google Scholar] [CrossRef]
- Frenkel, J. On the Transformation of light into Heat in Solids. I. Phys. Rev. 1931, 37, 17–44. [Google Scholar] [CrossRef]
- Koti, A.S.R.; Taneja, J.; Periasamy, N. Control of coherence length and aggregate size in the J-aggregate of porphyrin. Chem. Phys. Lett. 2003, 375, 171–176. [Google Scholar] [CrossRef]
- Romeo, A.; Castriciano, M.A.; Zagami, R.; Pollicino, G.; Monsu Scolaro, L.; Pasternack, R.F. Effect of zinc cations on the kinetics for supramolecular assembling and the chirality of porphyrin J-aggregates. Chem. Sci. 2017, 8, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerrum, J.; McReynolds, J.P.; Oppegard, A.L.; Parry, R.W. Hexamminecobalt(III) Salts. In Inorganic Syntheses; Mcgkatv-Hill Book Company, Inc.: New York, NY, USA, 1946; pp. 216–221. [Google Scholar] [CrossRef]
- Lee, C.S.; Gorton, E.M.; Neumann, H.M.; Hunt, H.R. Optically Active Tris(1,10-phenanthroline) Complexes of Chromium(III) and Cobalt(III) by Resolution and Synthesis. Inorg. Chem. 1966, 5, 1397–1399. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
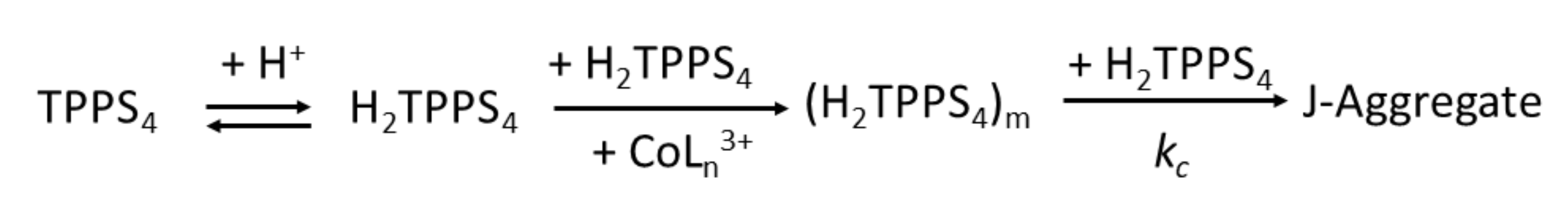{kind=link}
{kind=link}
| 104 × k0/ s−1 | 103 × kc/ s−1 | m | n | |
|---|---|---|---|---|
| [Co(NH3)6]3+ | ||||
| 100 | 0.65 ± 0.16 | 1.52 ± 0.01 | 3.2 ± 0.1 | 4.3 ± 0.2 |
| 200 | - | 3.00 ± 0.01 | 2.2 ± 0.1 | 2.2 ± 0.1 |
| 300 | 3.27 ± 0.98 | 8.44 ± 0.04 | 3.3 ± 0.1 | 2.9 ± 0.1 |
| [Co(phen)3]3+ | ||||
| 100 | 2.85 ± 0.17 | 0.863 ± 0.013 | 2.8 ± 0.1 | 3.0 ± 0.2 |
| 200 | 5.41 ± 0.27 | 2.46 ± 0.01 | 2.7 ± 0.1 | 2.7 ± 0.1 |
| 300 | 5.17 ± 0.37 | 4.38 ± 0.01 | 2.7 ± 0.1 | 2.6 ± 0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Manganaro, N.; Zagami, R.; Trapani, M.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M. Role of Cobalt(III) Cationic Complexes in the Self-Assembling Process of a Water Soluble Porphyrin. Int. J. Mol. Sci. 2021, 22, 39. https://doi.org/10.3390/ijms22010039
Manganaro N, Zagami R, Trapani M, Castriciano MA, Romeo A, Scolaro LM. Role of Cobalt(III) Cationic Complexes in the Self-Assembling Process of a Water Soluble Porphyrin. International Journal of Molecular Sciences. 2021; 22(1):39. https://doi.org/10.3390/ijms22010039
Chicago/Turabian StyleManganaro, Nadia, Roberto Zagami, Mariachiara Trapani, Maria Angela Castriciano, Andrea Romeo, and Luigi Monsù Scolaro. 2021. "Role of Cobalt(III) Cationic Complexes in the Self-Assembling Process of a Water Soluble Porphyrin" International Journal of Molecular Sciences 22, no. 1: 39. https://doi.org/10.3390/ijms22010039