The Westward Journey of Alfalfa Leaf Curl Virus

 , , , , ,
, , , , ,  , ,
, ,
Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Sampling
2.2. DNA Extraction and PCR-Mediated Alfalfa Leaf Curl Virus Detection
2.3. Cloning and Sequencing of ALCV Full Length Genomes
2.4. Pairwise-Distance, Phylogenetic, and Recombination Analyses
2.5. Statistical Analyses
3. Results and Discussion
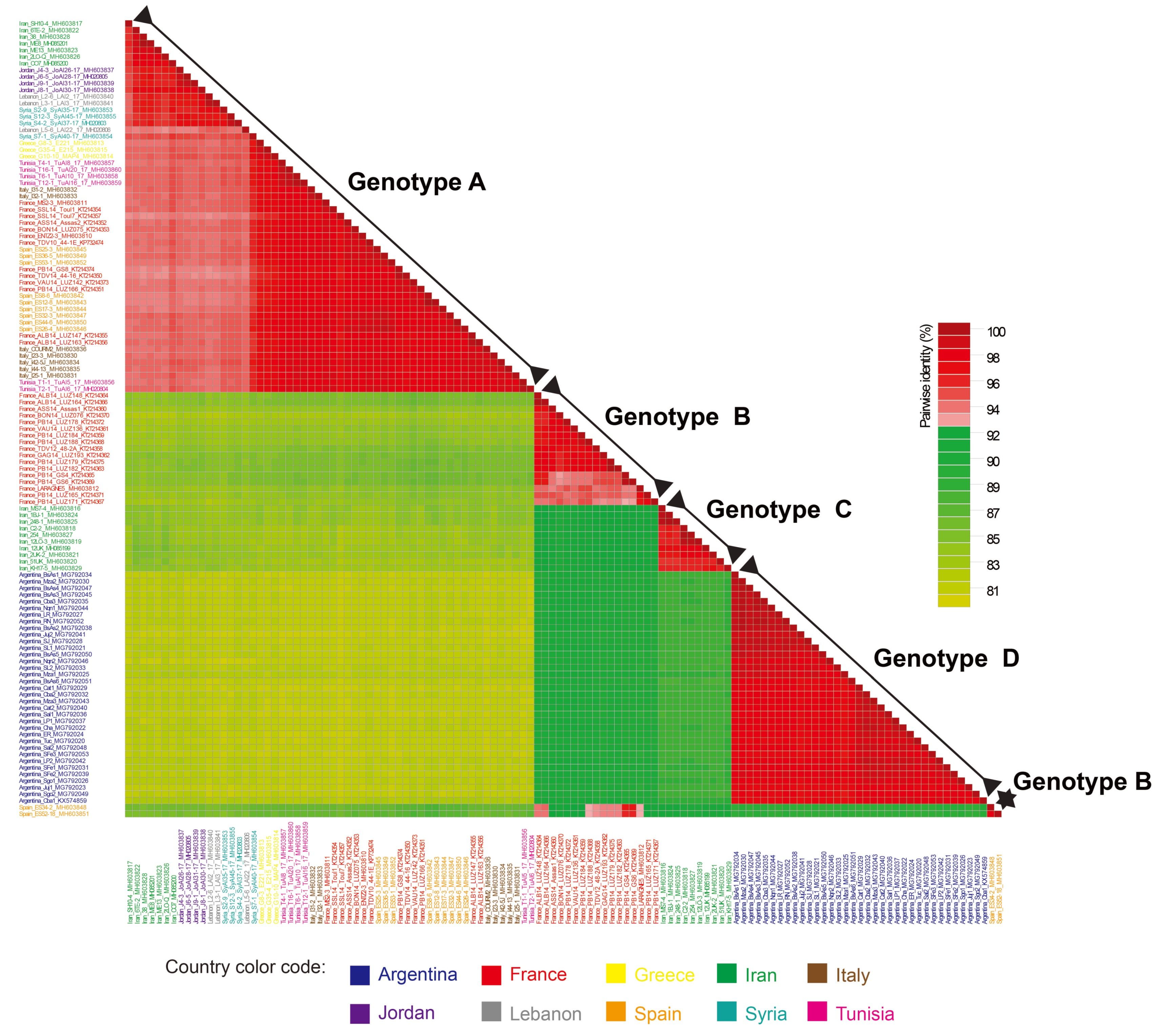
3.1. Genetic Diversity of ALCV Isolates
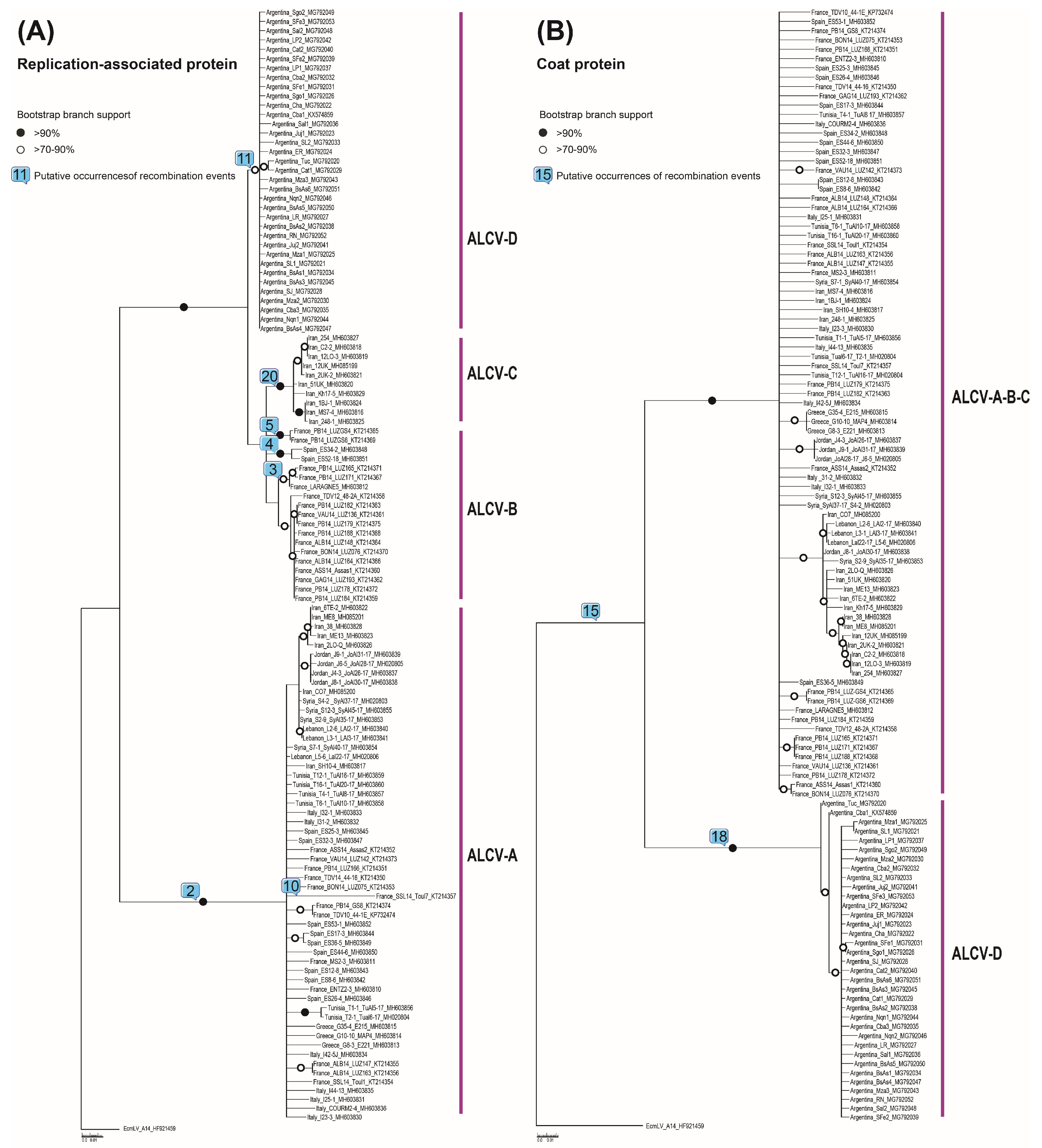
3.2. Phylogenetic and Recombination Analyses
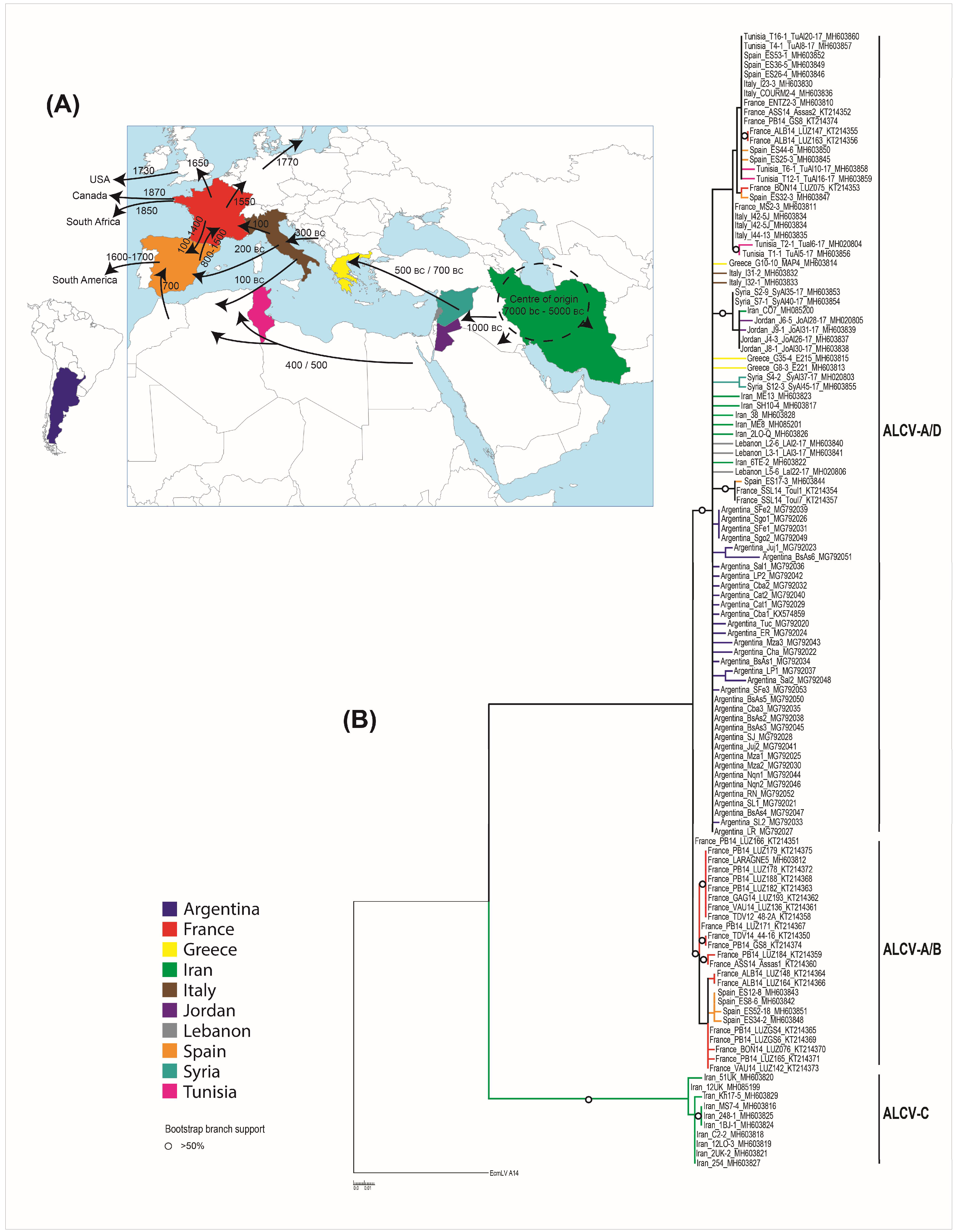
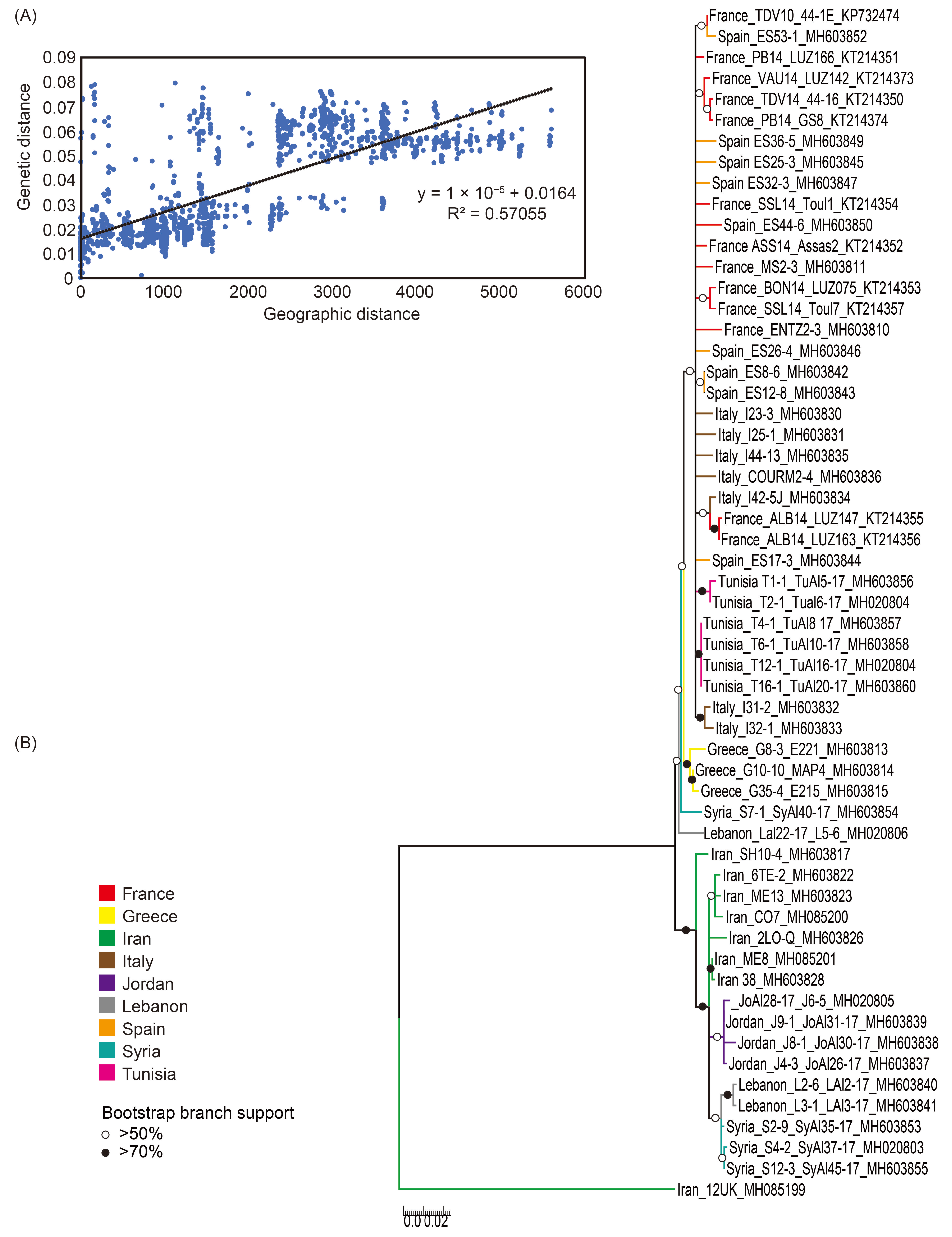
3.3. Geographic Distribution of ALCV
3.4. Geographical Origin of ALCV
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bock, K.R.; Guthrie, E.J.; Woods, R.D. Purification of maize streak virus and its relationship to viruses associated with streak diseases of sugarcane and panicum maximum. Ann. Appl. Biol. 1974, 77, 289–296. [Google Scholar] [CrossRef]
- Duffus, J.E.; Gold, A.H. Infectivity neutralization used in serological tests with partially purified beet curly top virus. Phytopathology 1973, 63, 1107–1110. [Google Scholar] [CrossRef]
- Claverie, S.; Bernardo, P.; Kraberger, S.; Hartnady, P.; Lefeuvre, P.; Lett, J.M.; Galzi, S.; Filloux, D.; Harkins, G.W.; Varsani, A.; et al. From spatial metagenomics to molecular characterization of plant viruses: A geminivirus case study. Adv. Virus Res. 2018, 101, 55–83. [Google Scholar] [PubMed]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant virus metagenomics: Advances in virus discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Roumagnac, P.; Fuchs, M.; Navas-Castillo, J.; Moriones, E.; Idris, A.; Briddon, R.W.; Rivera-Bustamante, R.; Zerbini, F.M.; Martin, D.P. Capulavirus and grablovirus: Two new genera in the family geminiviridae. Arch. Virol. 2017, 162, 1819–1831. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Golden, M.; Akram, M.; Naimuddin; Nadarajan, N.; Fernandez, E.; Granier, M.; Rebelo, A.G.; Peterschmitt, M.; Martin, D.P.; et al. Identification and characterisation of a highly divergent geminivirus: Evolutionary and taxonomic implications. Virus Res. 2013, 177, 35–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roumagnac, P.; Granier, M.; Bernardo, P.; Deshoux, M.; Ferdinand, R.; Galzi, S.; Fernandez, E.; Julian, C.; Abt, I.; Filloux, D.; et al. Alfalfa leaf curl virus: An aphid-transmitted geminivirus. J. Virol. 2015, 89, 9683–9688. [Google Scholar] [CrossRef] [PubMed]
- Susi, H.; Laine, A.L.; Filloux, D.; Kraberger, S.; Farkas, K.; Bernardo, P.; Frilander, M.J.; Martin, D.P.; Varsani, A.; Roumagnac, P. Genome sequences of a capulavirus infecting plantago lanceolata in the aland archipelago of finland. Arch. Virol. 2017, 162, 2041–2045. [Google Scholar] [CrossRef] [PubMed]
- Zerbini, F.M.; Briddon, R.W.; Idris, A.; Martin, D.P.; Moriones, E.; Navas-Castillo, J.; Rivera-Bustamante, R.; Roumagnac, P.; Varsani, A.; Ictv Report, C. ICTV virus taxonomy profile: Geminiviridae. J. Gen. Virol. 2017, 98, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Charles-Dominique, T.; Barakat, M.; Ortet, P.; Fernandez, E.; Filloux, D.; Hartnady, P.; Rebelo, T.A.; Cousins, S.R.; Mesleard, F.; et al. Geometagenomics illuminates the impact of agriculture on the distribution and prevalence of plant viruses at the ecosystem scale. ISME J. 2018, 12, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, P.; Muhire, B.; Francois, S.; Deshoux, M.; Hartnady, P.; Farkas, K.; Kraberger, S.; Filloux, D.; Fernandez, E.; Galzi, S.; et al. Molecular characterization and prevalence of two capulaviruses: Alfalfa leaf curl virus from france and euphorbia caput-medusae latent virus from south africa. Virology 2016, 493, 142–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumari, S.G.; Moukahel, A.R.; Richet, C.; Galzi, S.; Filloux, D.; Roumagnac, A.; Asaad, N.; Hijazi, J.; Mghandef, S. First report of alfalfa leaf curl virus affecting alfalfa (medicago sativa l.) in jordan, lebanon, syria and tunisia. Plant Dis. 2018, 102, 2052. [Google Scholar] [CrossRef] [PubMed]
- Davoodi, Z.; Heydarnejad, J.; Massumi, H.; Richet, C.; Galzi, S.; Filloux, D.; Roumagnac, A. First report of alfalfa leaf curl virus from alfalfa in iran. Plant Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bejerman, N.; Trucco, V.; de Breuil, S.; Pardina, P.R.; Lenardon, S.; Giolitti, F. Genome characterization of an Argentinean isolate of alfalfa leaf curl virus. Arch. Virol. 2018, 163, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Alliot, B.; Signoret, P.A.; Giannott, J. Presentation of bacilliform virus-particles associated with enation disease of alfalfa (medicago-sativa l). Cr. Acad. Sci. D Nat. 1972, 274, 1974–1976. [Google Scholar]
- Blattný, C. Virus papillosity of the leaves of lucerne. Folia Microbiol. 1959, 4, 212–215. [Google Scholar] [CrossRef]
- Cook, A.A.; Wilton, A.C. Alfalfa enation virus in the Kingdom of Saudi Arabia. FAO Plant Prot. Bull. 1984, 32, 139–140. [Google Scholar]
- Leclant, F.; Alliot, B.; Signoret, P.A. Transmission et épidémiologie de la maladie à énations de la luzerne (lev). Premiers résultats. Ann. Phytopathol. 1973, 5, 441–445. [Google Scholar]
- Rodriguez Sardiña, J.; Novales Lafarga, J. Una virosis de la alfalfa con produccíon de “enations”. An. INIA/Ser. Prot. Veg. 1973, 3, 132–146. [Google Scholar]
- Alliot, B.; Signoret, P.A. La “maladie à énations de la luzerne”, une maladie nouvelle pour la france. Phytopath. Z. 1972, 74, 69–73. [Google Scholar] [CrossRef]
- Annicchiarico, P. Alfalfa forage yield and leaf/stem ratio: Narrow-sense heritability, genetic correlation, and parent selection procedures. Euphytica 2015, 205, 409–420. [Google Scholar] [CrossRef]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Muhire, B.M.; Varsani, A.; Martin, D.P. Sdt: A virus classification tool based on pairwise sequence alignment and identity calculation. PLoS ONE 2014, 9, e108277. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murell, B.; Golden, M.; Khoosal, A.; Muhire, B. Rdp4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Stover, B.C.; Muller, K.F. Treegraph 2: Combining and visualizing evidence from different phylogenetic analyses. BMC Bioinform. 2010, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Posada, D. Prottest: Selection of best-fit models of protein evolution. Bioinformatics 2005, 21, 2104–2105. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. Genalex 6.5: Genetic analysis in excel. Population genetic software for teaching and research-an update. Bioinformatics 2012, 28, 2537–2539. [Google Scholar] [CrossRef] [PubMed]
- Fondong, V.N. Geminivirus protein structure and function. Mol. Plant Pathol. 2013, 14, 635–649. [Google Scholar] [CrossRef] [PubMed]
- Lima, A.T.M.; Silva, J.C.F.; Silva, F.N.; Castillo-Urquiza, G.P.; Silva, F.F.; Seah, Y.M.; Mizubuti, E.S.G.; Duffy, S.; Zerbini, M.F. The diversification of begomovirus populations is predominantly driven by mutational dynamics. Virus Evol. 2017, 3. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Holmes, E.C. Phylogenetic evidence for rapid rates of molecular evolution in the single-stranded DNA begomovirus tomato yellow leaf curl virus. J. Virol. 2008, 82, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Harkins, G.W.; Delport, W.; Duffy, S.; Wood, N.; Monjane, A.L.; Owor, B.E.; Donaldson, L.; Saumtally, S.; Triton, G.; Briddon, R.W.; et al. Experimental evidence indicating that mastreviruses probably did not co-diverge with their hosts. Virol. J. 2009, 6, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefeuvre, P.; Martin, D.P.; Harkins, G.; Lemey, P.; Gray, A.J.; Meredith, S.; Lakay, F.; Monjane, A.; Lett, J.M.; Varsani, A.; et al. The spread of tomato yellow leaf curl virus from the middle east to the world. PLoS Pathog. 2010, 6, e1001164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monjane, A.L.; Harkins, G.W.; Martin, D.P.; Lemey, P.; Lefeuvre, P.; Shepherd, D.N.; Oluwafemi, S.; Simuyandi, M.; Zinga, I.; Komba, E.K.; et al. Reconstructing the history of maize streak virus strain a dispersal to reveal diversification hot spots and its origin in Southern Africa. J. Virol. 2011, 85, 9623–9636. [Google Scholar] [CrossRef] [PubMed]
- Blackman, R.L.; Eastop, V.F. Aphids as Crop Pests. In Taxonomic Issues; van Emden, H.F., Harrington, R., Eds.; CABI: Wallingford, UK, 2007; pp. 1–29. [Google Scholar]
- CIE. Distribution Maps of Plant Pests; CAB International: Wallingford, UK, 1983. [Google Scholar]
- Coeur d’acier, A.; Jousselin, E.; Martin, J.F.; Rasplus, J.Y. Phylogeny of the genus aphis linnaeus, 1758 (homoptera: Aphididae) inferred from mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2007, 42, 598–611. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.P.; Morgan, D.J.; Havenstein, D.E. The ecology of Aphis craccivora koch and subterranean clover stunt virus. I. The phenology of aphid populations and the epidemiology of virus in pastures in South-East Australia. J. Appl. Ecol. 1971, 8, 699–721. [Google Scholar] [CrossRef]
- Anabestani, A.; Behjatnia, S.A.A.; Izadpanah, K.; Tabein, S.; Accotto, G.P. Seed transmission of beet curly top virus and beet curly top iran virus in a local cultivar of petunia in iran. Viruses 2017, 9, 299. [Google Scholar] [CrossRef] [PubMed]
- Kil, E.J.; Kim, S.; Lee, Y.J.; Byun, H.S.; Park, J.; Seo, H.; Kim, C.S.; Shim, J.K.; Lee, J.H.; Kim, J.K.; et al. Tomato yellow leaf curl virus (TYLCV-IL): A seed-transmissible geminivirus in tomatoes. Sci. Rep. 2016, 6, 19013. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kil, E.J.; Kim, S.; Seo, H.; Byun, H.S.; Park, J.; Chung, M.N.; Kwak, H.R.; Kim, M.K.; Kim, C.S.; et al. Seed transmission of sweet potato leaf curl virus in sweet potato (ipomoea batatas). Plant Pathol. 2015, 64, 1284–1291. [Google Scholar] [CrossRef]
- Prosperi, J.M.; Jenczewski, E.; Muller, M.H.; Fourtier, S.; Sampoux, J.P.; Ronfort, J. Alfalfa domestication history, genetic diversity and genetic resources. Legume Perspect. 2014, 13–14. [Google Scholar]
- Muller, M.H.; Poncet, C.; Prosperi, J.M.; Santoni, S.; Ronfort, J. Domestication history in the medicago sativa species complex: Inferences from nuclear sequence polymorphism. Mol. Ecol. 2006, 15, 1589–1602. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of ALCV Isolates | Averages of Genome-Wide Pairwise Identities (%) |
|---|---|
| Genotype A isolates (n = 56) | 95.7 |
| Genotype B isolates (n = 19) | 96.4 |
| Genotype C isolates (n = 10) | 97.1 |
| Genotype D isolates (n = 35) | 98.1 |
| Genotype A isolates (Middle East, n = 18) a | 96.4 |
| Genotype A isolates (Western Mediterranean basin, n = 38) b | 98.0 |
| Event | Recombinant(s) | Major Parent | Minor Parent | Methods a | Breakpoints Positions b |
|---|---|---|---|---|---|
| 1 | France_VAU14_LUZ142_KT214373 | France_PB14_GS4_KT214365 | Unknown | GBMST | 1420 (nad) c–2736 (nad) |
| 2 | Jordan_J9-1_MH603839 | EcmLV_A14_HF921459 | Argentina_Cba1_KX574859 | GBMCST | 1407 (nad)–2704 (nad) |
| 3 | France_PB14_LUZ171_KT214367 | Unknown | France_LARAGNE5_MH603812 | RGBMCST | 1332 (nad)–2193 |
| 4 | Spain_ES34-2_MH603848 | France_PB14_GS4_KT214365 | Iran_SH10-4_MH603817 | RGMCST | 2097 (nad)–2196 |
| 5 | France_PB14_GS4_KT214365 | France_ALB14_LUZ148_KT214364 | Spain_ES52-18_MH603851 | RBMCT | 1750 (nad)–2053 |
| 6 | France_BON14_LUZ076_KT214370 | France_GAG14_LUZ193_KT214362 | Unknown | RGBMCST | 434–1251 |
| 7 | France_PB14_LUZ184_KT214359 | France_TV12_48-2A_KT214358 | France_LARAGNE5_MH603812 | RGBMCST | 845–1027 |
| 8 | Iran_MS7-4_MH603816 | Iran_12LO-3_MH603819 | Iran_SH10-4_MH603817 | RMCST | 277–1295 (nad) |
| 9 | France_GAG14_LUZ193_KT214362 | France_TDV12_48-2A_KT214358 | Tunisia_T6-1_MH603858 | MST | 291–1261 |
| 10 | France_SSL14_Toul7_KT214357 | France_TDV10_44-1_KP732474 | Unknown | RGMCST | 2124–2355 |
| 11 | Argentina_Sal2_MG792048 | Unknown | Spain_ES52-18_MH603851 | RBMCT | 1259–2070 (nad) |
| 12 | Spain_ES52-18_MH603851 | Unknown | France_PB14_LUZ179_KT214375 | RGMCT | 289 (nad)–1333 |
| 13 | France_PB14_LUZ179_KT214375 | France_LARAGNE5_MH603812 | Tunisia_T2-1_MH020804 | RGMCST | 868–1333 (nad) |
| 14 | France_ASS14_Assas1_KT214360 | France_PB14_LUZ171_KT214367 | Italy_COURM2-4_MH603836 | RGMCST | 901 (nad)–1029 |
| 15 | France_TDV10_44-1_KP732474 | France_LARAGNE5_MH603812 | EcmLV_A14_HF921459 | RM | 872–1053 (nad) |
| 16 | France_PB14_LUZ171_KT214367 | France_PB14_LUZ179_KT214375 | Spain_ES34-2_MH603848 | RGS | 2434–2537 |
| 17 | France_LARAGNE5_MH603812 | France_VAU14_LUZ142_KT214373 | France_BON14_LUZ076_KT214370 | RMC | 844 - 1026 |
| 18 | Argentina_Mza3_ MG792043 | Unknown | France_LARAGNE5_MH603812 | MC | 211–1261 (nad) |
| 19 | Syria_S2-9_MH603853 | Iran_38_MH603828 | Spain_ES8-6_MH603842 | MC | 1797 (nad)–2093 |
| 20 | Iran_2UK-2_MH603821 | France_PB14_LUZ184_KT214359 | EcmLV_A14_HF921459 | RG | 1976 (nad)–2016 |
| 21 | Argentina_Nqn2_MG792046 | Argentina_BsAs1_ MG792034 | Unknown | RBT | 2493 (nad)–2565 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davoodi, Z.; Bejerman, N.; Richet, C.; Filloux, D.; Kumari, S.G.; Chatzivassiliou, E.K.; Galzi, S.; Julian, C.; Samarfard, S.; Trucco, V.; et al. The Westward Journey of Alfalfa Leaf Curl Virus. Viruses 2018, 10, 542. https://doi.org/10.3390/v10100542
Davoodi Z, Bejerman N, Richet C, Filloux D, Kumari SG, Chatzivassiliou EK, Galzi S, Julian C, Samarfard S, Trucco V, et al. The Westward Journey of Alfalfa Leaf Curl Virus. Viruses. 2018; 10(10):542. https://doi.org/10.3390/v10100542
Chicago/Turabian StyleDavoodi, Zohreh, Nicolás Bejerman, Cécile Richet, Denis Filloux, Safaa G. Kumari, Elisavet K. Chatzivassiliou, Serge Galzi, Charlotte Julian, Samira Samarfard, Verónica Trucco, and et al. 2018. "The Westward Journey of Alfalfa Leaf Curl Virus" Viruses 10, no. 10: 542. https://doi.org/10.3390/v10100542